|
����
ҽ����е��Ʒע���ϳ���֬���걨����Ҫ��
����(һ)��������
����1.����
����(1)���ݡ�ҽ����е����Ŀ¼��(����ʳƷҩƷ�ල�����ֹܾ���2017���104��)���ϳ���֬�����յ�����ҽ����е���й������������Ϊ17;
����(2)��Ʒ����Ӧ���ϡ�ҽ����еͨ��������������(����ʳƷҩƷ�ල�����ܾ����19��)���Ҫ���磺“�ϳ���֬��”��“��ϩ����֬��”�ȡ�
����2.��Ʒ����
�����ϳ���֬����Ʒ����Ӧȫ�桢��ϸ������Ҫ�����£�
����(1)������״����ѧ��ɳɷ�
����Ӧ������Ʒ������״��������Ʒ����̬���ߴ硢ɫ�ŵȣ���Ҫ��ʱ���ṩ��Ʒ��ģ��ͼ�ס�
����ҽ����еע��Ӧ������Ʒ����ɳɷּ����������������ʡ����ϡ�����������ɫ����ż�����ȳɷּ���������������ֵĻ�ѧ���ơ��ṹʽ�ͷ�������������Χ������ֵ����á�����ԭ���ϵ���Դ�����ϵı����ʿ�Ҫ��
����������ǿ��Ʒ��е���ܵĸ��Գɷ֣�Ӧ�����仯ѧ�ɷֵĵ�������ٷֺ�����������������(������)���ֲ��ȡ�
����(2)��������
����Ӧ������Ʒ�����ٴ�ʹ��Ҫ������߱��Ļ������ܣ������ߴ��ȶ��ԡ�����ݻ��е�ճ�����ܣ�ɫ����ȶ��ԣ����ѡ��������������ܣ��Լ�ʹ�ð�ȫ���ܷ����Ҫ��ͬʱӦ������Ʒ����Ҫ�������ϵı���
����(3)��ƷӦ�ü���
����Ӧ�����ϳ���֬����Ʒ���ٴ�Ӧ�ü���������Ӧ�úϳ���֬�����оֲ�����ȫ��������������У��Բ�Ʒʹ�ò���Ӱ��IJ������أ��������ϳ���֬���ͺš�ɫ�ŵ�ѡ����ʱĥͷ��ѡ�����Ĵ�����ʽ�����������ѡ��ʹ�÷�ˮ��ƾ������ľ�ʾ�ȡ�
����3.�ͺŹ��
������ͬһע�ᵥԪ�ڣ����齫���в�ͬ��νṹ����ͬ���νṹ����ͬɫϵ�IJ�Ʒ����Ϊ��ͬ�ͺţ�����Ҫ��Ҳ�ɽ�������������(��)����װ(28��)���а�װ���;���������ͬ��νṹ�����νṹ��ɫϵ����ͬ�ߴ�IJ�Ʒ����Ϊ��ͬ������걨��Ʒ���ڶ����ͺŵ����Σ���ȷ�������ݣ����Խṹʾ��ͼ������˵�����ϵ���ʽ������ͬ�ͺŲ�Ʒ�����νṹ���������岢��ʾ�ؼ��ߴ硣
����4.��Ʒ��װ˵��
����Ӧ��ͼƬ���������ϵķ�ʽ��ʾ�걨��Ʒ�İ�װ��Ϣ�����б���ʽ˵�����а�װ������ṩ��װ���ϵ����ʣ����۰�װ���϶Բ��ϴ�����̵�Ӱ�졣������Ʒ��װװ����
����5.���÷�Χ�ͽ���֢
�����ϳ���֬�������ڿ�ǻ���оֲ���ݺ�ȫ����ݵ������������˿ɸ����걨��Ʒ�ľ���Ԥ����;���о����ϣ��ο���ָ��ԭ���������Ҫ���һ��ȷ���걨��Ʒ��������÷�Χ������֢��
�������÷�Χ�ı���Ӧ�ۡ�������ʹ������ȷ�������ҵ���ϵ���������ʣ�����Ӧ������Ʒ�������������������͡�
��������֢Ӧ�����ò�Ʒ�����õļ�����������ض�����Ⱥ���磺��ϩ����������߽��õȡ�
����6.��ͬ���Ʒ��ǰ����Ʒ�ıȽ���Ϣ
����Ӧ�ṩͬ���Ʒ(������������)��ǰ����Ʒ(����)����Ϣ����������ע���Ʒ���з�������Ŀ�ġ�����ͬ���Ʒ��Ӧ��˵��ѡ������Ϊ�з��ο���ԭ��
����Ӧ����ͬ���Ʒ�������о����ٴ�ʹ����״����չ���ơ�ͬʱ�б��Ƚ�˵����Ʒ��ο���Ʒ(ͬ���Ʒ��ǰ����Ʒ)�ڲ��Ͻṹ��ɡ������ա�����ָ�ꡢӦ�ü����ȷ������ͬ��
����(��)�����
�����ϳ���֬����Ʒ��ɳɷ֡���������С���ۺϷ�ʽ�ͼӹ�����(����ͼ���)�Ⱦ���Ӱ�����ղ�Ʒ���ܣ���Ʒ�о���������Ӧ�����������ݣ�
����1.ԭ���Ͽ���
������ȷ��Ʒ����ʼ���ʣ�������Ʒ��������������ʼ�������ղ�Ʒ�ӹ�����������ȫ�����ϣ����������ʳɷ֡�����������ۼ������ϡ�����������ɫ����ż������ȫ��ԭ���ϵĻ�ѧ���ơ�CAS�š���ѧ�ṹʽ/����ʽ������������ֲ�����Դ�ʹ���(������)��ʹ��������ɱ��������ϵı��������˵����ձ�����صİ�ȫ�����۱��棬�������б�����ʽ�ṩ��
���������״����ڴ����Ʒ���²��ϣ�Ӧ�ṩ�ò����ʺ�����Ԥ�����÷�Χ�İ�ȫ�ԡ���Ч������о�����(�����������ڸóɷֵĻ�ѧƷ���ϰ�ȫ���ݱ��Լ����ԡ��°��ԡ���ͻ���ԡ��̼���������֤�ݵ�)��
����2.����ԭ������ɳɷ��о�
����(1)�ϳ���֬����Ʒ�ɷַ�����������Ӱ���Ʒ��������ܡ�Ӳ�ȵ����ܡ�Ӧ�ṩ��ɳɷ־ۺ�����ϵķ�������������Χȷ������;
����(2)���Ӿ����洦����SiO2������������Ϻ����Ʒ�ϸ�Ӳ�Ⱥ���ĥ�Եģ�Ӧ��ȷ��ǿ�����͡����ϻ������û���;�����������ٷֺ���/����ٷֱȵ�ȷ������;�������ڸ߾����еķ�ɢ�븴��״̬;�����϶Բ��ϵļӹ�����Ӱ���;
����(3)���ý����ľۺϵ����������ľۺ������ӣ� �磺MPM(Premium��Heraeus Kulzer)��IPN(Trubyte IPN)��DCL(SR Postaris DCL)���γ��ڲ�������״��ͨ�ṹ����ǿ��е���ܵģ�Ӧ��ȷ����ԭ���������ۺ�����ӷ������ṹ���ṩ��ؼ�����ѧ���о�����;
����(4)�������������ģ������CNCϵͳ��������Ԥ�ɵ���֬���ϻ��߲��ù�̻�����Һ̬��֬���ϵ�3D��ӡ���������ģ�Ӧ��ȷ�ӹ�ԭ��(SLA��FDM��DLP��SLS��)����������ԭ������֡����ͺ������֣��ṩ��ؼ�����ѧ���о����ϡ�
����3.���ͷ����о�
�����ϳ���֬���Ĵ�ͳ�ӹ����ͷ�����Ҫ��ע�ܷ���ģѹ�����֣�Ӧ�ṩ���ͷ���ȷ�����ݡ��磺ģѹ���������̲�����ͬ��Ӧ�����������̺����ԡ�ע�ܷ������֬�����Ե�Ҫ����Ҫͨ���Ծۺ���ƽ�����������������ֲ��Ȳ����ĸı�������֬�ļӹ����ܼ��Կ��ƣ�Ӧ��������������Ի�������ԶԳ��ͺ���֬������Ӱ���ṩ�о����ϡ�
��������CNCϵͳ��������Ԥ�ɵ���֬���ϻ��߲��ù�̻����͵�3D��ӡ����������Ӧ��������õ���Ӳ��ϵͳ���ӹ����̡��ӹ����ȡ���е���ܺͱ�����ȵ���֬���ؼ�����ָ���Լ��������Ʒ������ṩ�о����ϡ�
����4.��Ʒ�����о�
����ҽ����еע�����������Ӧ���ݲ�Ʒ����ص��ύ�й����ܵ��о����ϣ������������ܡ���ѧ���ܡ����������Լ�������������ص�����ָ��ȷ�����ݣ��ṩ�����ñ������鷽�������ۻ�����ʵʩ���ݡ��������ٰ������������о���
����(1)���ijߴ�
�����ϳ���֬���ijߴ���������ģ��ͼ�����涨����ֵ���Ӧ������5%��
����(2)ɫ���ں���
����ǰ���ͺ�����ɫ�����������ı�ɫ���ָ����ɫ��Ƚϣ�Ӧ�����Բ��졣��ɫ���������Ե�;���֮�䲻Ӧ�����Եķֽ��ߡ�
����ע����Ҫ�����Ϊģ������ı�Ե����Ȼ�����ʵ���̬��ר����Ƶķֽ��ߡ�
����(3)�������
����Ŀ���۲죬����(��λ���ֳ���)�ı���Ӧ�⻬���й��������ס�
�����ϳ���֬��Ӧ�ܱ���ָ���ԭ�еĹ��ȡ�
����(4)��϶������ȱ��
�����ںϳ���֬�������ڱ����ϣ�Ӧ��϶��ȱ�ݣ�����ĥ�����ֲڵ���ĥ�ۼ������ۿɼ������ʡ�
����(5)����ݻ��оۺ����ճ������
����ҽ����е��Ʒע������ϳ���֬��Ӧ�������YY 0270.1-2011������ѧ ���оۺ��� ��1���֣���ݻ��оۺ��(ע����ָ��ԭ���б��������°汾����ͬ)����������ݻ��оۺ���(����)ճ���ι̡����չ涨�����鷽�����飬6�������У�������5�������ĸ��ղ�λ����ݻ��оۺ���ճ���ι̡������������ʽʵ������ݻ��оۺ���ճ�ӹ�λ�ģ�Ӧ����YY/T 0519-2009�����Ʋ��� �����ݽṹճ�ӵIJ��ԡ�����ع��ұ�����ҵ�����������ݵ�Ҫ��
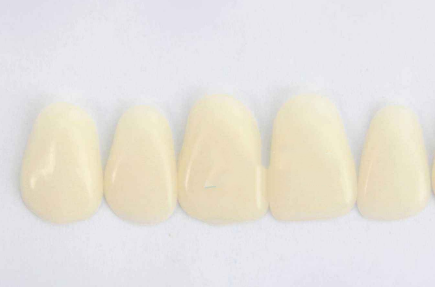
����(6)�����ס������Ρ�����
�������ݲ�Ӧ������Σ������ղ�λ��ֱ����Ե�ߵ����������ⲻӦ���ѡ�
����(7)ɫ���ȶ���
�������ݵı����䲿λ��û�б����䲿λ�Լ�δ�������֮�����ɫ��Ӧ�пɲ���仯��
����(8)�ߴ��ȶ���
������ֻ���Ľ�Զ�о��ߴ�仯��ΧӦ��ԭ�ߴ��±2%���ڡ�
����(9)Ӳ��
����Ӳ���DZ����ϳ���֬����Ʒ���ԡ����ԡ�ǿ�Ⱥ����Ե�һϵ�в�ͬ��������ϵ�һ���ۺ�����ָ�ꡣ���ϳ���֬��Ӳ�ȵͣ��ֿ�������Ӳ����ѹ��������������ײ�������ȱ��ȱ��;���ϳ���֬��Ӳ�ȹ���ҧ��ײ�������壬���������ѹ���ݼ������������յ����ա���ƷӲ��Ӧ�ں��������䷶Χ�ڣ���ͬʱ������Ȼ��Ӳ�ȡ��ϳ���֬����Ʒ��Ӳ�Ȳ����������Ŭ��ѹ�۷��ȡ�
����(10)��ĥ������
��������ҽ����еע����ĥ������Ӱ��ϳ���֬����Ʒ�ٴ�Ӧ��Ч������Ҫ���ܡ����ۺϳ���֬����ĥ���Եķ�����Ҫ���ٴ��۲����ۺ�����ģ��ʵ�飬����ģ��ʵ��ϳ����õķ���Ϊ���������ĥ��ʵ�顣�������YY/T 0113-2015 ������ѧ ������֬��ĥ�����ܲ��Է���������ɲ��á����ʱ�����֯/�����淶ISO/TS14569-1�����Ʋ��� ��ĥ������ָ�� ��һ����:��ˢ��ĥ�ġ��������ʱ�����֯/�����淶ISO/TS14569-2�����Ʋ���—��ĥ������ָ�� �ڶ����� ����/����Ӵ���ĥ�𡷷������С����������������Ӧ�ṩ������ѧ�����ݡ�������ǿ�ͺϳ���֬����������֬����ĥ����Ӧ���ڴ�ͳ�ۼ���ϩ������ϳ���֬����
����(11)��Ⱦɫ��
���������ƺϳ���֬����Ʒ���п�Ⱦɫ�ԣ�Ӧ�ύ��Ⱦɫ���о����ϡ���Ⱦɫ�Կ�ʹ�÷ֹ��ȱ�ɫ�ǣ������˹���ɫ�ȡ����������������Ӧ�ṩ������ѧ�����ݡ�
����(12)�������庬��
�����ϳ���֬�����ݲ�ͬ�ۺ���������ȷ���ۺ��ﵥ��ijɷ֣�������ѹ���͵ĺϳ���֬�����������ϩ��������庬�����ֵӦΪ2.2%(��������)��
��������������������Ʒ�IJ���庬����������Ҫ�������庬���ⶨֵӦ����������������ֵ+0.2%(��������)�����ⶨֵ����������������ֵ��0.2%֮�͡�
����(13)��������
������Ʒ���Ƶ��������ܣ��翹����Եȡ�
����5.����ҽ����е��Ʒע����������������
�����ϳ���֬�������������(��ǻճĤ)�־ýӴ��IJ�Ʒ����������������Ӧ��ѭGB/T 16886.1-2011��ҽ����е����ѧ���۵�1���֣����չ��������е����������顷��YY/T 0268-2008������ѧ ��ǻҽ����е����ѧ���� ��1��Ԫ�����������顷����ҽ����е����ѧ���ۺ����ָ�ϡ�(��ʳҩ��е�z2007�{345��)���Ҫ�������������������Ͽ��Կ���(������������)���·��棺
����(1)ҽ����е����ѧ���۵IJ��Ժ���������
����(2)ҽ����е���ò���ѡ�������
����(3)���ϱ���
����-ҽ����е���ϵĶ����붨����˵�������
����-ҽ����е���������۲�Ʒ�ĵ�ͬ�ԱȽ�
����(4)ѡ����������ѧ��������ɺ���֤
����(5)�������ݺ��������Ļ���
����(6)�������ѧ�����������������
������������ѧ����������ĿӦ����YY/T 0268-2008������ѧ ��ǻҽ����е����ѧ���� ��1��Ԫ�����������顷ȷ�������鿼�ǣ�
����ϸ������
����������
��������ȫ������
�����̼���Ƥ�ڷ�Ӧ
�����Ŵ�����
����������ȫ������
����6.��Ʒ��Ч�ںͰ�װ�о�
������Ʒ��Ч����ָ��Ʒ��һ�����¶ȡ�ʪ�ȡ����ߵ�������Ӱ���£���������������ѧ�����ʵ����ޡ��ϳ���֬����Ч����֤һ�������Ʒ��Ч����֤�Ͱ�װ��֤��
����(1)��Ʒ��Ч����֤���ṩ��Ʒ��Ч�ڼ���Ч����֤���ϡ�
������Ʒ��Ч����֤�������й����õĹ��ڡ����ʱ�����֤�������У��ύ��Ʒ����Ч����֤���档��Ʒ��Ч�ڿɲ���ʵʱ�ϻ��о��������ϻ��о���֤��
��������Ч���о��У�Ӧ���������Ч����ȷ����Ʒ��ȫ�Ժ���Ч�ԵĹؼ��������ڲ�Ʒ����Ҫ�����������IJ���(��ߴ硢�����ס������Ρ����ѡ�ɫ���ȶ��Ե�)�����ύ��ѡ����Է�������֤���ϡ�
������ѡ��ʵʱ�ϻ���Ч����֤���飬��Ʒѡ����ϻ���������Ӧ�����ڲ�Ʒ˵�������Ƶ����桢���价��������
������ѡ������ϻ���Ч����֤���飬Ӧ˵�����ü��������ĺ����ԡ����磬�ڱ��¶Ⱥ������¶�����µĽ������Ӧ���ǵ�Ч�ģ����¶ȸı��Arrhenius���ߵ�б�ʱ��ֲ��䡣�ڲ���֤ʵ��Ч��ʱ������ͬ�¶��¿��ɲ�ͬ���������ƷʧЧ��Ӧ�ύ����ĺ�����˵�����ڽ��м����ϻ������о�ʱӦע�⣺��Ʒѡ��Ļ����������ϻ�����Ӧ�����Ƶ����桢���价�������²�Ʒ�ϻ��Ļ�����ƥ�䡣
����(2)��װ��֤�������Ƶ���Ч�����Լ����䴢�������£����ְ�װ�����Ե����ݡ�
������Ʒ��װ��֤�������й����õĹ��ڡ����ʱ�����֤�������У��磺��Ʒ��װ�ĵ������顢�����顢�����¶����顢����ʪ������ȣ��ύ��Ʒ�İ�װ��֤���档��װ���ϵ�ѡ���鿼���������أ���װ���ϵ�������ѧ����;��װ�������Ʒ����Ӧ��;��װ��������ͺ��ܷ���̵���Ӧ��;��װ���������ṩ����������ѧ����;��װ�������ǩϵͳ����Ӧ��;��װ����������������̵��ʺ��ԡ���װ��֤����������Ӧ���װ˵���и�������Ϣ��������ڲ����õ�����Ӧ���������õ����ݡ�
�������ڰ�װ����Ч����֤��ҽ����еע����ѯ�����������ύ���ճ�Ʒ����װ�ij�ʼ�����Ժ�ά�������Ե���֤�����
����7.����֤����Ʒ��ȫ�ԡ���Ч�Ե��о�����
��������ܣ��ṩ�ϳ���֬����ҧ�Ϲ�ϵԭ�������ݡ���������ҧ�Ϲ�ϵ�����ϼ���άģ������ͼ��
����Ϊʹ�ϳ���֬������ݻ��оۺ����ι�ճ�ӣ���Ҫ������ӹ�����(��ĥ��)ʱ��Ӧ�ṩ���˵����
����(��)����������Ϣ
����1.ҽ����е��Ʒע���ύ��Ʒ���������չ��������ļ�����ϸ˵����Ʒ��ԭ��������Ʒ��ȫ�����������պͲ��裬�г�����ͼ����Ӧ��������·�ߡ��ؼ���������ա���������ָ�꼰�����֤���档 ��ȷԭ���Ʊ�����ɫ�����ϡ�Ͷ�ϡ���Һ��Ϲ��̡����ݡ�ģѹ��ע�ܡ��ȴ�����ȥ�����Ρ����Ρ����塢ģ��ά���ȵ��������Ƶ㼰��������ָ�꣬�ṩ��ص���֤���棬���Թ��յĿɿ��ԡ��ȶ��Խ���ȷ�ϡ���ȷ���������и��ָ��ϵ�ʹ����������ϲ�������Σ�����������Ʒ�����
�����ϳ���֬���Ĺؼ�����Ϊ���Ϲ��գ������Ϊ�����գ�Ŀǰ��������Ҫ����ģѹ����ע�ܷ���ģѹ���ǽ���״��ԭ�����ڱպ�ģǻ�ڽ������ȡ���ѹ���ۺϳ���Ϊ��Ʒ�ļӹ�����������ģѹ����������ȷ�¶ȡ�ѹ���Լ��ۺ�ʱ��;ע�ܷ���ָ��һ���¶Ⱥ�ѹ���£���ԭ��ͨ����ѹ����ģǻ�������ۺϺ�õ���Ʒ�ķ�����ע�ܷ���������ȷע��ѹ����ע���¶ȡ�ע��ʱ�䡢��ѹѹ���ͱ�ѹʱ��ȡ�
��������CNCϵͳ��������Ԥ�ɵ���֬���ϵģ�Ҫ��ȷCNCϵͳ����������������������(3��������4��������5������)������ֱ������ȴ��ʽ(���䡢ˮ�䡢��ȴҺ)��ϵͳ�ӹ����ȵȹؼ����������ù�̻�����Һ̬��֬���ϵ�3D��ӡ���������ģ�Ҫ��ȷ�ֲ��ȡ��ڷŽǶȡ�����(����ۺ�)ʱ���������������֧����Ʒ���������������ӡ·�������������Լ�ϵͳ�ӹ����ȵȹؼ�������
������������ҵ��ȡ�����ӹ������գ�����ݹ��ձ����ص���ȷ��Ӧ�Ĺ��ղ�����
����2.��������
����ҽ����еע�����걨��Ʒ�ж�����ơ��������أ�Ӧ����ÿ�����ơ��������صĸſ���
����(��)�ٴ���������
����������Ӧ����ҽ����еע������취��(����ʳƷҩƷ�ල�����ܾ����4��)����ҽ����е�ٴ����ۼ���ָ��ԭ��(����ʳƷҩƷ�ල�����ܾ�ͨ��2015���14��)ѡ��������ٴ����۷�ʽ���ύ�ٴ��������ϡ�
����1.���ݡ����ڹ����������ڽ����ٴ�����ҽ����еĿ¼��ͨ�桷(����ҩƷ�ල������ͨ��2018���94��)�����ڷ���YY 0300-2009������ѧ �����˹����������Ҫ�������ھֲ���ݺ�ȫ����ݵ����������ڽ����ٴ����顣�������������ʹ�����²��ϡ����Գɷ֡��¼���������ƻ���������û������¹��ܵIJ�Ʒ��
����2.�ٴ��������Ҫ��
�������й����ڿ�չ�ٴ������Ӧ���ϡ�ҽ����е�ٴ��������������淶��(����ʳƷҩƷ�ල�����ܾ� �л��������������ͼ���ίԱ�����25��)����ָ��ԭ���Ҫ��Ӧ�Բ�Ʒ�Ƿ�����ʹ��Ҫ��������÷�Χ����ȷ�ϡ��ύ���ٴ���������Ӧ�������ٴ�����Э�顢�ٴ����鷽�����ٴ����鱨��ȡ�
�������ݺϳ���֬��ʹ���ص㣬��Ʒ����Ч����ͨ���ϳ���֬������������ݵ��������������֤�����������辭���ٴ�������Ʋ��������Ҽӹ����������ϳ���֬������Ч�Բ���ȡ���ڲ��ϱ������ԣ����������������ƺͼӹ������йأ���˽���ϳ���֬�����ٴ�������ʹ�õ���������Ӧ����������ٴ�������ƺ淶�ļ����Ҳ�����Ҫ��
�����ٴ�����ʱӦע�����¼����棬���ο���
����(1)���Զ���
����ҽ����еע��������ݱ���ƷĿ��������Ⱥ�����ٴ��о������й涨���ƺ�������������������ų�����
���������߿�ǻ״������Ӧ��������ǻ����״�����������ȼ�ɫ������״����ҧ���������������ǻ�Ĥ���������Ⱦ����Ѫ����֢�ȸ��ֲ���״������֯ȱ�����ͼ��̶ȣ�����ȱ��ȱʧ�����Ԥ���IJ�λ��������������ʹ�ø�����������ȡ�
����(2)������ѡ�������������
�����ϳ���֬�����ٴ����������Ҫ�����������в�Ʒ��Ϊ�����飬���龡���ܲ�������������飬����ȷ�Ƚ�����(��Ч���顢��Ч�����Ч�����)�������ڷ���Ч�Լ��顢��Ч�Լ������Ч�Լ���ıȽ����ͣ�Ӧ���ȹ涨�����ٴ�����Ľ�ֵ������õ���Ŀ��ֵ����Ӧ������ȷĿ��ֵ����ȷ�����ݡ�
����(3)����ָ��
����������ָ��
����Ϊ���۱��Բ�Ʒ�����ܣ�Ӧ��ȷ�ٴ��������Ҫ��Ч����ָ�ꡢ��Ҫ��Ч����ָ�꼰��ȫ������ָ�ꡣ
������Ч������ָ���趨��������ĥ���ԡ����䡢�������ԡ���������(��ɫ)���Զ������ĥ�����ƽ�ȶȡ�ҧ�Ϲ�ϵ�ָ��ȡ��������ͺϳ���֬������Ӧ���������Ƶ���Ӧ֢�����ܵ�������Ӧ��������Ŀ��
������ȫ������ָ���趨���������ݺϳ���֬����Ʒ���ԣ���Ʒ�Կ�ǻ�Ĥ��֯�Ĵ̼��ԡ���ǻ��֯��ȫ���Ĺ������Ϊ��������ָ�ꡣ�磺������Ӧ�������¼�������֢�����Ƽ�顢���������ȡ�
���������۱�
��������ָ������ù��ʹ��ϵ����۱���������ϵı���������ٴ�������Ч���۱���
�����ٴ���������У���Ӧ��¼���黼�ߵľ�������Ͳ���ʱ�䣬��¼���ٴ��о��ڼ��κο���Ӱ����������ҩ�����Ƽ�����/ʹ�ü������翹���ء���ʹ��������ˮ�ȵ���ҩ����ȡ�
����(4)�ٴ��۲�
��������ǰ����
������ǰ����Ӧ�����������ߵ�ȫ��״��������Ӱ�����������κμ�������������߿�ǻ״����������Ԥ���IJ�λ���������ͼ��̶ȡ���������͡�����Ĥ����������������ܽ���״�������۹����ճ̶ȵȡ�
�������ٴ���������
����Ӧ��ϸ��¼�ٴ��IJ������裬��������ٴ���Ʒ���������ļ����Ҽӹ����輰����������������ʹ�õ��������ϵȡ�
��������������
���������ٴ����۱����йص���Ŀ��������м������ۣ�����¼���۵Ľ����
�������ٴ��۲�ʱ��
����ҽ����е��Ʒע��������ݺϳ���֬���ص�ȷ�����ʵ��ٴ��۲켰���ʱ�䡣�����ٴ�����۲�ʱ���Ϊ��������1�ܡ�1���¡�3���¡�6���¡������������εĽ����ʵ��ӳ��ٴ�����۲�ʱ�䣬�磺i.�ϳ���֬����ɲ���ȱ���ٴ�Ӧ�õİ�ȫ����;ii.ȱ���걨��Ʒ�ٴ��۲�����(6����)�����������ȶ�������;iii.Ӧ�ü����������еĺϳ���֬����Ʒ��ͬ;iiii.������Ϊ��Ҫ�ӳ��ٴ�����۲�ʱ������Ρ�
����(5)����������
�����ɸ�������������ȷ�����������ƵIJ���������
���������ȡ�������������;
���������ȡ�ıȽ����ͣ���Ч���顢��Ч�����Ч����;
�����������������Դ���ĸ���a(ͨ��������˫��0.05)�ͷ������Դ���ĸ���b(ͨ��������0.2);
��������Ҫ����ָ������ͺ��йص�ЧӦ��С�������̶�;
����������ǵ�Ч�����Ч���飬Ӧ�ṩ���ٴ�����Ľ�ֵ;
��������ʵ�������ȷ�����ܵIJ��������ʣ��Ա�֤���㹻�İ��նȼ�������졣
����(6)ͳ�Ʒ���
����Ӧ��������������͡��Ƚ����͡��������ʺ�ͳ�Ʒ���Ŀ�ģ�����ѡ��ͳ�Ʒ�����������ȷ����ͳ�Ʒ������ݼ����弰ͳ�Ʒ���������
����ͳ�Ʒ�������Ӧ���ٰ��������IJ��֣�
�������ٴ����������������������ٴ�����ſ�(ɸѡ�������������������������ʧ��/�˳�/��������);
�����ڻ���������Ӧ��������ѡ������(ITT������)�Ļ����˿�ͳ��ѧָ�꼰������ز�ʷָ��Ƚ���ͳ������;
��������Ч/Ч�����ۣ�Ӧ��������ѡ��������(ITT������)��������������������(PP������)�ֱ����ͳ�Ʒ����������۽����һ���ԡ���Ч����ʱ����������⣬��Ӧ��������Ƶ�95%�������������;
�����ܰ�ȫ������ʱ��Ӧ��������ѡ�������߽��з���(SS������)��������©���з������κβ����¼��������з����IJ����¼�Ӧ�������Ƿ������о���Ʒ�йء�
����(7)�ٴ���������
�����ٴ����鱨��Ӧ���ٴ����鷽������һ�£�����ע����ȷ�������ݣ������Ʒ�����ƣ�����ͺţ��������ͣ�����Ӧ�ò�λ�������������ʱ�䣬�����Ʒ���ٴ���Ӧ֢������֢��ע�����
�����ٴ����鱨��������ȷ���в����Ƿ�ȫ�������ã������ò����Ƿ������ͳ�ƣ�ʧ�ò�������ȷʧ��ԭ��
�����ٴ����鱨�������ύ������Ч�����밲ȫ�����۵�ͳ�ƹ��������漰����ԭʼ���ݡ�
�����ٴ����鱨�����豨�����в�����Ӧ�Ͳ����¼�������ʱ�䡢������ԭ������������ò�Ʒ�Ĺ�ϵ����������ȡ�Ĵ�ʩ��������ȷ��
�����ٴ����鱨��Ӧ���о���λ����ͳ�Ʒ������棬������ȷ���ٴ�������ۡ�
����(��)��Ʒ�ķ��շ�������
����ҽ����е��Ʒע�ᰴ��YY/T 0316-2016��ҽ����е ���չ�����ҽ����е��Ӧ�á�����Ҫ�Բ�Ʒ��������ȫ����ʵʩ���չ������������ڲ�Ʒ��ע������ǰ��Ӧ�Է��չ������̽�����������Ӧ����ȷ������Ʒ�ķ����ѱ�ȫ��ط���;���չ����ƻ��ѱ��ʵ���ʵʩ;�ۺ�ʣ������ǿɽ��ܵ�;�����ʵ�������������������������Ϣ��������Ӧ�γɷ��չ������档���չ�������Ӧ���ٰ���������Ϣ��
����1.����Ӱ���Ʒ��ȫ�Ե����������嵥
��������ҽ����еע��������Ӧ�ο�YY/T 0316-2016��¼C��Ҫ���ж�ҽ����е�밲ȫ���й����������⣬��ʶ����յ���Դ���������ڴˡ�������Ӧ�Ը����Ʒ���г�ֵķ���ʶ�𣬷���ʶ�����Ϣ��Դ��Ҫ�����г����ɰ�����������������;�������Ʋ�Ʒ��Ͷ��/��Թ���ݡ�ҽѧ���ס�ʵ���Ҽ�⡢��Ʒ��ǩ��ʶ��ר�ҹ۵�ȡ����ڷ���ʶ����Ϣ����Դ��ҵӦ����˵�������ύ�й�֧���ļ������ס�
����2.��Ʒ�й�Σ�����嵥
����������Ӧ��ϸ�г����Ʒ�йص���֪�Ϳ�Ԥ��Σ�����嵥���Լ���ÿ��Σ���������ķ���(������Ԥ�����¼����С�Σ�������Ϳ��ܷ�������)��
����������Ӧָ�����걨��Ʒ�����е��κζ�����գ�˵�����շ����ķ�������ʶ��ķ���Ӧ���ٰ����������������·��棺
����(1)���ϵ�����ѧ�ͻ�ѧΣ����
�������ϵĻ�ѧ�ṹ����Դ
�������ϵ�����������
����(2)�����ӹ����̿��ܲ�����Σ����
������Ⱦ
�������Ӽ�(����)�IJ���
�������������ྻ��
����(3)��Ʒʹ�÷������أ�
�����̼��ԡ������ԡ�����ʧЧ
����(4)����ȷʹ�ò�����Σ����
����ʹ�ò�Ʒʱδ����˵�����в�����������
��������˵���������÷�Χ������֢����ʾ��Ϣ����
����(5)��Ʒ��װ���ܲ�����Σ����
������װ����
������ʶ����
�����������������̲�����
��������ҽ����еע��֤������Ӧ����ʶ��ķ����������Ľ��ͷ��յĴ�ʩ���������걨��Ʒ�ķ���Ӧ����YY/T 0316-2016Ҫ�����δ���ơ�������˵������п��ǡ�
����������Ӧ�ڲ�Ʒ����ȫ�����жԷ��ս��й������ƣ���ʹʣ������ڿɽ��ܷ�Χ�ڡ���ͨ����Ʒ��ƿ��ơ���Ʒԭ����ѡ��Ʒ��������ָ����ƶ����ٴ����顢��ȷ�ı�ǩ��ʶ�������ͼ�����ơ���Ʒ˵����ȶ����ʩ�Խ��ͷ������ɽ���ˮƽ����������������������
����(��)��Ʒ����Ҫ��
����������Ӧ��ϲ�Ʒ�ļ����������ٴ���Ʒ�ص㣬����������Ч�Ĺ��ұ�����ҵ�����л�����ҩ�䡷(���¼�ơ��й�ҩ�䡷)��ȷ����Ʒ��ȫ��Ч�������ɿص�����ָ������鷽������Ʒ����Ҫ����Ӧ��ȷ����ͺż��仮�ֵ�˵������Ʒ������һ����Ϣ(ԭ���ϡ���ɳɷּ��ٷֺ�������νṹ�����Ρ��ߴ��)����Ʒ����ָ�꼰���鷽�����Բ�Ʒ����Ҫ�������е�������Ŀ�����õ����鷽�����м�Ҫ�����������ù��ϱ��е����鷽������ֱ�����ø÷�����������Ӧ���ı�š���ż����й�ҩ�䡷�İ汾�š����Թ��ϱ��е����鷽�������ģ���Ӧ˵���ĵ����ݼ�ԭ��ÿһ��������Ŀ����������������ʽ�������ؽ����������б��������������������Ľ��ܱ���
�����ƶ��ϳ���֬������Ҫ��ij��òο������£�
����YY 0300-2009������ѧ �����˹�����
������Ʒ����Ҫ���е�����ָ��Ӧ������YY 0300-2009�е����Ҫ���鷽��Ӧ������ҵ���еķ���������������������Ӧѡ����֤�ķ�����˵��ԭ��
����ҽ����е��ѯ��ָ��ԭ������˺ϳ���֬��������ָ��Ҫ���鿼�ǵ��������������ݣ�
����(1)���ijߴ�
����(2)ɫ���ں���
����(3)�������
����(4)��϶������ȱ��
����(5)����ݻ��оۺ����ճ������
����(6)�����ס������Ρ�����
����(7)ɫ���ȶ���
����(8)�ߴ��ȶ���
�������в����õ���Ŀ��Ӧ����˵�������½��ṩ�˳����Ʒ�Ļ�������Ҫ����ο���
������������Ϊ“Ӳ�ʺϳ���֬��”ʱ��Ӧ��ȷӲ��ָ�꼰�����鷽����������Ӧ���о���ͬʱӦ��˵��������ȷ��
��������3D��ӡ��CAD/CAM���յĺϳ���֬��������ʽ��֬���Ȳ�ƷӦ���ݲ�Ʒ�����ص��ƶ���Ӧ������ָ�꣬Ӧ�����ڷ���YY 0300-2009������ѧ �����˹����������Ҫ����ʹ���²��ϡ��¼���������ƻ���������û������¹��ܵIJ�Ʒ�������������ܼ��������Ƶ���������Ҫ��Ӧ�ڼ���Ҫ������ȷ��
����(��)��Ʒע����鱨��
������Ʒ��ע����Ӧ�ھ���ҽ����е�������ʵļ���������У���ƷӦ�ڼ�������м췶Χ�ڡ�������Ӧ�ṩҽ����е����������ߵļ��鱨���Ԥ������������⣬��Ӧ�ṩ������Ʒ����ͺŵ�ѡ�����ݡ��������ͺŲ�ƷӦ���DZ�ע�ᵥԪ���ܹ������걨�������ͺŲ�Ʒ��ȫ�Ժ���Ч�Եĵ��Ͳ�Ʒ��
�����ϳ���֬���ĵ�����ѡ��Ӧ���ǻ�ѧ��֡������ա��ṹ�ߴ�����أ�ѡ��������ȫ���ṹ��ӡ�������ߵIJ�Ʒ�����ͺż�IJ���Բ�Ʒ���ܺͼ�����������Ӱ�죬Ӧ�ֱ�ѡȡ������Ʒ����ȫ���ܼ���;Ҳ�ɸ��ݲ�������ѡ������ͺŽ���ȫ���ܼ��飬ѡ�������ͺŽ��в����Լ��飬�磺ɫ��/��ɫ��ͬ�ĺϳ���֬������ɫ��/��ɫ��ͬ������ɫ���ɷֲ�ͬ���£��ɿ��ǶԲ�ͬɫ��/��ɫ��Ʒ����ɫ��ɫ���ȶ��ԵȽ��м�⡣
����(��)˵����ͱ�ǩ����
������Ʒ˵����ͱ�ǩ�ı�дӦ���ϡ�ҽ����е˵����ͱ�ǩ�����涨��(����ʳƷҩƷ�ල�����ܾ����6��)��Ҫ�����ύ���ı��ͱ�ǩ��ͼӦ����������������˵�����������������÷�Χ��Ӧ�ü���������֢��Ӧ���Ʒ���ٴ����۱���һ�¡���Ʒ���������ṹ��ɡ�������Ч�ڵ�Ӧ���������Ϻ��о�����������������֤������һ�¡�
����˵���鲻Ӧ����û�����ݺͿ���˵IJ�Ʒ����ָ�꣬Ӧʵ�����ǵؽ��ܲ�Ʒ���ص㡣�����Ʋ�Ʒ���ܺ��ص�ģ�Ӧ�в�Ʒ����Ҫ����ύ��ע�Ἴ���������ݡ���ʹ��“ϵ��”��“��”��“��”��“��������”�Ⱥ���;������ôʡ�
�������⣬��Ӧ�����������ݵ������
����1.�ϳ���֬������Ҫ�ɷּ��京��;
����2.Ϊ�ﵽ�ϳ���֬������ݻ��оۺ����ι�ճ�ӣ���Ҫ�������(����ĥ)ʱ��Ӧ��˵��������ϸָ��;
����3.��Ҫʱ��Ӧ�ṩ��ɫ�塢ģ��ͼ�ס�
���ں�Զҽ����е��ѯ����˾��һ�Ҽ���רҵ��ҽ����е��ѯ����˾��רע�ṩȫ�������磺���ڡ����ݡ���ݸ����ɽ����ɽ�����ݡ�˳�¡��Ϻ������������졢�ɶ���֪�����е�ҽ����е��������ѯ����רҵ��ҽ����е��Ʒע��֤������ҽ����е��Ʒ����綨��������ҽ����е��������֤������ҽ����е��Ӫ����֤������ҽ����е��Ӫ����������ҽ����еע�ᡢһ��ҽ����е��Ʒ����������������FDAע�ᡢISO13485��֤�� CE��֤������������������֤���ٴ����顢����֤����������֤�ȴ�������ҽ����е����������ϵ��֤�ļ��Ľ�������ϵ�����ȷ���ļ��Ľ��� (�磺ISO9001�� ISO13485 ��GMP�� CE��QSR820��CMDCAS)��Ʒ��⣬�ٴ����鼰���ٴ����ϱ�д����Ʒ����Ҫ���ƶ��������ļ���д��������ż������ġ�ҽ����е�����������������ྻ�ҽ���ָ���ȷ�����רҵ�����ŷ��������õͣ���ӭ����ѯ��
����
|

