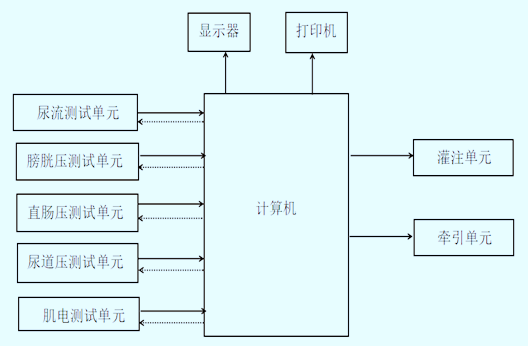
|
标准编号 |
标准名称 |
|
GB 9706.1—2007 |
医用电气设备 第1部分:安全通用要求 |
|
GB 9706.15—2008 |
医用电气设备 第1-1部分:通用安全要求
并列标准:医用电气系统安全要求 |
|
GB/T 191—2008 |
包装储运图示标志 |
|
GB/T 14710—2009 |
医用电器环境要求及试验方法 |
|
GB/T 20271—2006 |
信息安全技术信息系统通用安全技术要求 |
|
GB/T 25000.51—2016 |
系统与软件工程 系统与软件质量要求和评价(SQuaRE) 第51部分:就绪可用软件产品(RUSP)的质量要求和测试细则 |
|
YY 0505—2012 |
医用电气设备 第1-2部分:安全通用要求并列标准电磁兼容要求和试验 |
|
YY 0896—2013 |
医用电气设备 第2部分:肌电及诱发反应设备安全专用要求 |
|
YY/T 0316—2016 |
医疗器械风险管理对医疗器械的应用 |
|
YY/T 0466.1—2016 |
医疗器械用于医疗器械标签、标记和提供信息的符号 第1部分:通用要求 |
|
YY/T 0664—2008 |
医疗器械软件 软件生存周期过程 |
|
YY/T 0708—2009 |
医用电气设备第1-4部分:安全通用要求:可编程医用电气系统 |
|
YY/T 1095—2015 |
肌电生物反馈仪 |
|
YY/T 1406.1—2016 |
医疗器械软件 第1部分:YY/T 0316应用于医疗器械软件的指南 |