|
ЎЎЎЎ
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІбТЅУГїШОВМєЈ¬ёГАаІъЖ·НЁ№эїШЦЖЙи±ёДЪС»·ТєМеµДОВ¶ИЈ¬ѕЯУР¶ФИЛМеЅшРРМеНвОпАнЙэОВєН/»тЅµОВ№¦ДЬЈ¬ґпµЅёЁЦъµчЅЪИЛМеОВ¶ИДїµДµДЙи±ёЈ¬УГУЪЛДЦ«єН¶оН·Ад/ИИ·уµДІъЖ·ЎЈДЗГґТЅУГїШОВМєІъЖ·ЧўІбЦ¤ЙуЕъјјКхЧКБПИзєО±аРґ?¶јУРДДР©ТЄЗуДШ?ѕЯМеЗлїґТЅБЖЖчРµТЅУГїШОВМєІъЖ·ЧўІбјјКхЧКБПЙк±Ё·Ѕ°ёТЄЗуЈє
ЎЎЎЎ(Т»)ІъЖ·ГыіЖТЄЗу
ЎЎЎЎ1.ІъЖ·ГыіЖЅЁТй№ж·¶ОЄ“ТЅУГїШОВМє”ЎЈ
ЎЎЎЎ2.ИфІъЖ·ЅцѕЯУРЅµОВ№¦ДЬЈ¬ІъЖ·ГыіЖЅЁТй№ж·¶ОЄ“ТЅУГЅµОВМє”Ј¬ИфІъЖ·ЅцѕЯУРЙэОВ№¦ДЬЈ¬ІъЖ·ГыіЖЅЁТй№ж·¶ОЄ“ТЅУГЙэОВМє”ЎЈ
ЎЎЎЎ(¶ю)ТЅБЖЖчРµЧўІбІъЖ·µДЅб№№єНЧйіЙ
ЎЎЎЎ°ґїШОВДїµД·ЦОЄЈєµҐАдРНЎўµҐИИРНЎўАдИИРНЎЈ
ЎЎЎЎ°ґЧйіЙ·ЦОЄЈєЦч»ъ(ИзНј1ЛщКѕ)ЎўМєЧУєНМеОВґ«ёРЖчµИЎЈ
ЎЎЎЎ°ґПµНі·ЦОЄЈєїШОВПµНіЧйјюЎўїШЦЖПµНіЧйјюЎўЛ®С»·ПµНіЧйјюєНїЗМеЧйјюµИЎЈ
ЎЎЎЎ(Иэ)ТЅБЖЖчРµІъЖ·ЧўІбґъАнІъЖ·№¤ЧчФАнєНЧчУГ»ъАн
ЎЎЎЎ1.ІъЖ·№¤ЧчФАн
ЎЎЎЎїШОВМє№¤ЧчФАнЈєФЪЦч»ъ№©Л®їЪУл»ШЛ®їЪЙПЅУЙПДЪУРС»·№ЬВ·µДМєЧУЈ¬ЦРСлїШЦЖЖчНЁ№эИЛМеОВ¶ИїШЦЖ·ґАЎ¶ФС№Лх»ъЎў·зЙИЎўЛ®±ГµИЅшРРКµК±їШЦЖЈ¬јґїЙКµПЦМєЧУµДС»·Л®ЦЖАдЎўЦЖИИµДОВ¶ИїШЦЖЈ¬С»·Л®Ул»јХЯ·ўЙъИИБїЅ»»»Ј¬ґпµЅїШЦЖМеОВДїµДЈ¬ИзНј2ЛщКѕЎЈ
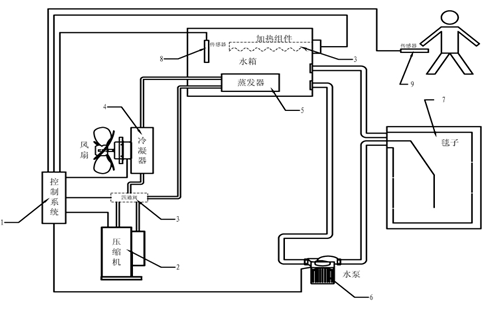
ЎЎЎЎ1.їШЦЖПµНі;2.С№Лх»ъ;3.ЛДНЁ·§»тјУИИЧйјю(ЦЖИИ№¦ДЬК±їЙСЎФсЛДНЁ·§»тјУИИЧйјю);4.АдДэЖч;5.Хф·ўЖч;6.Л®±Г;7.МєЧУ;8.Л®ОВґ«ёРЖч;9.МеОВґ«ёРЖчЎЈ
ЎЎЎЎ2.ІъЖ·ЧчУГ»ъАн
ЎЎЎЎТтёГІъЖ·ОЄ·ЗЦ±ЅУЦОБЖАаТЅБЖЖчРµЈ¬№К±ѕЦёµјФФтІ»°ьє¬ІъЖ·ЧчУГ»ъАнµДДЪИЭЎЈ
ЎЎЎЎ(ЛД)ЧўІбµҐФЄ»®·ЦµДФФтєНКµАэ
ЎЎЎЎТЅБЖЖчРµЧўІбґъАнТЅУГїШОВМєµДЧўІбµҐФЄФФтЙПТФјјКхЅб№№ЎўРФДЬЦё±кЎўФ¤ЖЪУГНѕЧчОЄ»®·ЦЧўІбµҐФЄµДТАѕЭЎЈ
ЎЎЎЎІ»Н¬µДµз»ч·А»¤АаРНУ¦ЧчОЄІ»Н¬ЧўІбµҐФЄЅшРРЧўІбЎЈИзµз»ч·А»¤АаРН·Ц±рОЄIАаЎўўтАаїШОВМєЈ¬У¦°ґХХБЅёцЧўІбµҐФЄЅшРРЧўІбЎЈ
ЎЎЎЎµҐАдРНЎўµҐИИРНј°АдИИРНїШОВМєУ¦°ґХХІ»Н¬µДТЅБЖЖчРµЧўІб°мАнµҐФЄЅшРРЧўІбЎЈ
ЎЎЎЎ(Ое)ІъЖ·ККУГµДПа№Ш±кЧј
ЎЎЎЎёщѕЭІъЖ·ЧФЙнМШµгККУГ±н1ЦРПа№Ш±кЧјЈє±кЧј±аєЕ±кЧјГыіЖ
ЎЎЎЎGB/T 191—2008°ьЧ°ґўФЛНјКѕ±кЦѕ
ЎЎЎЎGB/T 9969.1—2008№¤ТµІъЖ·К№УГЛµГчКйЧЬФт
ЎЎЎЎGB 9706.1—2007ТЅУГµзЖшЙи±ёµЪ1Ії·ЦЈє°ІИ«НЁУГТЄЗу
ЎЎЎЎGB/T 14710—2009ТЅУГµзЖч»·ѕіТЄЗуј°КФСй·Ѕ·Ё
ЎЎЎЎGB/T 16886.1—2011ТЅБЖЖчРµЙъОпС§ЖАјЫµЪ1Ії·ЦЈє·зПХ№ЬАн№эіМЦРµДЖАјЫУлКФСй(ИзККУГ)
ЎЎЎЎGB/T 16886.5—2003ТЅБЖЖчРµЙъОпС§ЖАјЫµЪ5Ії·ЦЈєМеНвПё°ы¶ѕРФКФСй(ИзККУГ)
ЎЎЎЎGB/T 16886.10—2005ТЅБЖЖчРµЙъОпС§ЖАјЫµЪ10Ії·ЦЈєґМј¤УліЩ·ўРНі¬Гф·ґУ¦КФСй(ИзККУГ)
ЎЎЎЎYY/T 0316—2016ТЅБЖЖчРµ·зПХ№ЬАн¶ФТЅБЖЖчРµµДУ¦УГ
ЎЎЎЎYY 0505—2012ТЅУГµзЖшЙи±ёµЪ1—2Ії·ЦЈє°ІИ«НЁУГТЄЗуІўБР±кЧјЈєµзґЕјжИЭТЄЗуєНКФСй
ЎЎЎЎYY 0709—2009ТЅУГµзЖшЙи±ёµЪ1—8Ії·ЦЈє°ІИ«НЁУГТЄЗуІўБР±кЧјЈєНЁУГТЄЗуЈ¬ТЅУГµзЖшЙи±ёєНТЅУГµзЖшПµНіЦР±ЁѕЇПµНіµДІвКФєНЦёДП(ИзККУГ)
ЎЎЎЎYY 0785—2010БЩґІМеОВјЖБ¬РшІвБїµДµзЧУМеОВјЖРФДЬТЄЗу
ЎЎЎЎYY 0834—2011ТЅУГµзЖшЙи±ёµЪ2Ії·ЦЈєТЅУГµзИИМєЎўµзИИµжєНµзИИґІµж°ІИ«ЧЁУГТЄЗу
ЎЎЎЎYY 0952—2015ТЅУГїШОВМє
ЎЎЎЎЙПКц±кЧј°ьАЁБЛІъЖ·јјКхТЄЗуЦРѕіЈЙжј°µЅµДІїјю±кЧјєН·Ѕ·Ё±кЧјЎЈУРµДЖуТµ»№»бёщѕЭІъЖ·µДМШµгТэУГТ»Р©РРТµНвµД±кЧјєНТ»Р©ЅПОЄМШКвµД±кЧјЎЈ
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІбґъАнІъЖ·ККУГј°ТэУГ±кЧјµДЙуІйїЙТФ·ЦБЅІЅАґЅшРРЎЈКЧПИ¶ФТэУГ±кЧјµДЖлИ«РФєНККТЛРФЅшРРЙуІйЈ¬ТІѕНКЗФЪ±аРґІъЖ·јјКхТЄЗуК±УлІъЖ·Па№ШµД№ъјТЎўРРТµ±кЧјКЗ·сЅшРРБЛТэУГЈ¬ТФј°ТэУГКЗ·сЧјИ·ЎЈїЙТФНЁ№э¶Ф“·ыєПРФЙщГч”ЦРЙщГч·ыєПµДПа№Ш±кЧјКЗ·сЖлИ«ЎўККТЛАґЅшРРЙуІйЎЈґЛК±Ј¬У¦ЧўТв±кЧј±аєЕЎў±кЧјГыіЖКЗ·сНкХы№ж·¶Ј¬ДкґъєЕКЗ·сУРР§ЎЈЖдґО¶ФТэУГ±кЧјµДІЙДЙЗйїцЅшРРЙуІйЎЈјґЛщТэУГµД±кЧјЦРµДМхїоТЄЗуЈ¬КЗ·сФЪІъЖ·јјКхТЄЗуЦРЅшРРБЛКµЦКРФµДМхїоТэУГЎЈ
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІб°мАнЙПКц±кЧјИзУРРВ°ж·ўІјКµК©Ј¬У¦ЦґРРЧоРВ°ж±ѕЎЈ
ЎЎЎЎ(Бщ)ІъЖ·µДККУГ·¶О§/Ф¤ЖЪУГНѕЎўЅыјЙЦў
ЎЎЎЎ1.ТЅУГїШОВМє(АдИИРН)
ЎЎЎЎККУГ·¶О§Т»°гОЄ“ККУГУЪТЅБЖ»ъ№№ёЯИИ»јХЯОпАнЅµОВєНµНОВ»јХЯОпАнЙэОВТФј°РиТЄ±ЈіЦМеОВµД»јХЯ”ЎЈ
ЎЎЎЎ2.ТЅУГЅµОВМє(µҐАдРН)
ЎЎЎЎККУГ·¶О§Т»°гОЄ“ККУГУЪТЅБЖ»ъ№№ёЯИИ»јХЯОпАнЅµОВТФј°РиТЄ±ЈіЦМеОВµД»јХЯ”ЎЈ
ЎЎЎЎ3.ТЅУГЙэОВМє(µҐИИРН)
ЎЎЎЎККУГ·¶О§Т»°гОЄ“ККУГУЪТЅБЖ»ъ№№µНОВ»јХЯОпАнЙэОВТФј°РиТЄ±ЈіЦМеОВµД»јХЯ”ЎЈ
ЎЎЎЎЅыјЙЦўЈєФЭОґ·ўПЦЎЈ
ЎЎЎЎ(ЖЯ)ІъЖ·µДЦчТЄ·зПХј°СРѕїТЄЗу
ЎЎЎЎТЅБЖЖчРµЧўІбґъ°мТЅУГїШОВМєµД·зПХ№ЬАн±ЁёжУ¦·ыєПYY/T 0316—2016Ў¶ТЅБЖЖчРµ·зПХ№ЬАн¶ФТЅБЖЖчРµµДУ¦УГЎ·µДУР№ШТЄЗуЈ¬ЙуІйТЄµг°ьАЁЈє
ЎЎЎЎ1.ІъЖ·УР№ШµД°ІИ«РФМШХчЕР¶ЁїЙІОїјYY/T 0316—2016µДёЅВјCЎЈ
ЎЎЎЎ2.ОЈє¦ЎўїЙФ¤јыµДКВјюРтБРєНОЈє¦ґ¦ѕіЕР¶ПїЙІОїјYY/T 0316—2016ёЅВјEЎўIЎЈ
ЎЎЎЎ3.·зПХїШЦЖµД·Ѕ°ёУлКµК©ЎўЧЫєПКЈУа·зПХµДїЙЅУКЬРФЖАјЫј°ЙъІъєНЙъІъєујаКУПа№Ш·Ѕ·ЁїЙІОїјYY/T 0316—2016ёЅВјFЎўGЎўJЎЈ
ЎЎЎЎ4.·зПХїЙЅУКЬЧјФтЈ¬ЅµµН·зПХµДґлК©ј°ІЙИЎґлК©єу·зПХµДїЙЅУКХіМ¶ИЈ¬КЗ·сУРРВµД·зПХІъЙъЎЈ
ЎЎЎЎТЅБЖЖчРµЧўІбґъАнПВ±нТАѕЭYY/T 0316—2016µДёЅВјE(±нE.1)БРѕЩБЛїШОВМєІъЖ·УР№ШµДїЙДЬОЈє¦КѕАэµДІ»НкИ«З嵥Ј¬ТФ°пЦъЕР¶ЁУлїШОВМєІъЖ·УР№ШµДОЈє¦ЎЈЖуТµ»№У¦ёщѕЭЧФЙнІъЖ·МШµгИ·¶ЁЖдЛыїЙДЬОЈє¦ЎЈХл¶ФІъЖ·µДёчПо·зПХЈ¬ЖуТµУ¦ІЙИЎїШЦЖґлК©Ј¬И·±Ј·зПХЅµµЅїЙЅУКЬµДіМ¶ИЎЈ
ЎЎЎЎ±н2 ОЈє¦АаРНЎўРОіЙТтЛШј°·А·¶їШЦЖґлК©
ЎЎЎЎОЈє¦АаРНРОіЙТтЛШ
ЎЎЎЎДЬБїОЈє¦µзґЕДЬїЙґҐј°ЅрКфЎўНвїЗµИУлґшµзІї·ЦёфАл/±Ј»¤І»№»Ј¬µзЅйЦКЗї¶ИІ»№»Ј¬їЙДЬ¶ФК№УГХЯФміЙµз»чОЈє¦ЎЈ
ЎЎЎЎІъЖ·НвїЗѕшФµ/ёфАлІ»№»Ј¬їЙДЬТэЖр№эБїВ©µзБчЙЛє¦К№УГХЯ»т»јХЯЎЈ
ЎЎЎЎї№µзґЕёЙИЕДЬБ¦ІоЎўМШ¶Ё»·ѕіПВ№¤ЧчІ»ХэіЈЈ¬»тёЙИЕЖдЛыЙи±ёХэіЈ№¤ЧчЎЈ
ЎЎЎЎИИДЬОґ°ІЧ°Л®В·і¬ОВ±Ј»¤Ўўґ«ёРЖч№КХП±Ј»¤МбКѕµИЧ°ЦГЈ¬µјЦВ№эёЯ»т№эµНОВ¶ИКдіцЈ¬їЙДЬТэЖр»јХЯММЙЛ»т¶іЙЛЎЈ
ЎЎЎЎ»ъРµДЬІъЖ·ГжЎўЅЗЎў±ЯґЦІЪЈ¬¶јїЙДЬ¶ФК№УГХЯ»т»јХЯФміЙ»ъРµЛрЙЛЎЈ
ЎЎЎЎЙъОпС§єН»ЇС§ОЈє¦ЙъОпС§єН
ЎЎЎЎ»ЇС§ОЈє¦ІъЖ·ЗеЅа»тПы¶ѕІ»НкИ«Ј¬їЙДЬ»бК№»јХЯЖ¤·фёРИѕЈ¬ПёѕъЎўІЎ¶ѕµИЅшИл»јХЯМеДЪЎЈ
ЎЎЎЎЙъОпПаИЭРФУ¦УГІї·ЦИфЦ±ЅУУл»јХЯЖ¤·фЅУґҐЈ¬МєГжІДБПУ¦ЅшРРЙъОпПаИЭРФЖАјЫЎЈ
ЎЎЎЎІЩЧчОЈє¦К№УГґнОуИХіЈК№УГЎўО¬»¤ЎўРЈЧјОґ°ґ№ж¶ЁЅшРРЈ¬µјЦВІъЖ·Ж«АлХэіЈК№УГЧґМ¬ЎЈ
ЎЎЎЎРЕПўОЈє¦І»ККµ±µД±кјЗ±кјЗИ±ЙЩ»тІ»ХэИ·Ј¬±кјЗµДО»ЦГІ»ХэИ·Ј¬І»ДЬ±»ХэИ·µШК¶±рЈ¬І»ДЬУАѕГМщАОєНЗеіюТЧИПµИЎЈ
ЎЎЎЎІ»НкХыµДЛµГчКйЛµГчКйЦР¶ФІъЖ·РФДЬМШХчЎўФ¤ЖЪУГНѕЎўК№УГПЮЦЖµИГиКцІ»№ж·¶ЎўІ»НкХыЈ¬µјЦВІъЖ·µД·ЗФ¤ЖЪ»ті¬·¶О§К№УГЎЈ
ЎЎЎЎІ»ККµ±µДІЩЧчЛµГчИХіЈК№УГЎўО¬»¤ЎўРЈЧј№ж¶ЁІ»ГчИ·ЎўІ»ККµ±ЎЈ
ЎЎЎЎ(°Л)ТЅБЖЖчРµЧўІбЦ¤ЙкЗлІъЖ·јјКхТЄЗуУ¦°ьАЁµДЦчТЄРФДЬЦё±к
ЎЎЎЎІъЖ·јјКхТЄЗуЙуІйКЗІъЖ·ЦчТЄРФДЬЦё±кЙуІйЦРЧоЦШТЄµД»·ЅЪЦ®Т»ЎЈІъЖ·јјКхТЄЗуЦРµДјјКхЦё±кІї·ЦКЗ·сЖлИ«Ј¬їЙТФНЁ№э¶ФКЗ·сѕЯУРТФПВЦчТЄДЪИЭАґЅшРРЙуЖАЈє
ЎЎЎЎ1.ХэіЈ№¤ЧчМхјю
ЎЎЎЎїШОВМєµД№¤ЧчМхјюУЙЦЖФмЙМ№ж¶ЁЎЈ
ЎЎЎЎ2.РФДЬ
ЎЎЎЎ2.1С»·ТєМеОВ¶И
ЎЎЎЎ2.1.1С»·ТєМеОВ¶ИЙи¶Ё·¶О§УЙЦЖФмЙМ№ж¶ЁЎЈІЅЅшЈє≤1ЎжЎЈ
ЎЎЎЎ2.1.2С»·ТєМеОВ¶ИФКІоЈє±1.5ЎжЎЈ
ЎЎЎЎ2.2МеОВґ«ёРЖч
ЎЎЎЎ2.2.1їШОВПµНіїЙТФЙи¶Ё»јХЯµДДї±кМеОВЎЈЦЖАдЙи¶Ё·¶О§Јє30.0Ўж—40.0Ўж;ЦЖИИЙи¶Ё·¶О§Јє30.0Ўж—37.0ЎжЎЈІЅЅшЈє≤0.5ЎжЎЈ
ЎЎЎЎ2.2.2МеОВґ«ёРЖчјаІв·¶О§І»РЎУЪ28Ўж—43ЎжЈ¬ФКІоЈє±0.2ЎжЎЈ
ЎЎЎЎ2.3їХФШЖЅѕщЛЩВК
ЎЎЎЎЦЖАд/ЦЖИИїХФШЖЅѕщЛЩВКУ¦ФЪЦЖФмЙМ№ж¶ЁµД·¶О§ДЪ;ёГ·¶О§єН¶ФУ¦µДОВ¶И±д»ЇЗшјдУ¦УЙЦЖФмЙМ№ж¶ЁЎЈ
ЎЎЎЎ2.4ёєФШЧоґуЖЅѕщЛЩВК
ЎЎЎЎФЪ№ж¶ЁµДёєФШМхјюПВЈ¬ЦЖАд/ЦЖИИЧоґуЖЅѕщЛЩВК·¶О§УЙЦЖФмЙМ№ж¶ЁЎЈ
ЎЎЎЎ2.5ФлЙщ
ЎЎЎЎїШОВМєХэіЈ№¤ЧчК±Ј¬ФлЙщ≤60dB(A)ЎЈ
ЎЎЎЎ2.6іРЦШТЄЗу
ЎЎЎЎїШОВМєХэіЈ№¤ЧчК±Ј¬МєЧУіРЦШУ¦≥135kgЎЈ
ЎЎЎЎ2.7МєЧУіЯґз
ЎЎЎЎУЙЦЖФмЙМ№ж¶ЁЈ¬ФКРнОуІо±5%ЎЈ
ЎЎЎЎ2.8ГЬ·вРФ
ЎЎЎЎїШОВМєС»·№ЬВ·ГЬ·вУ¦БјєГЈ¬ОЮР№В©ПЦПуЎЈ
ЎЎЎЎ3.№¦ДЬ
ЎЎЎЎ3.1С»·ТєМеОВ¶Иі¬№э42ЎжК±Ј¬У¦НЈЦ№№¤ЧчЈ¬ІўѕЯУРМбКѕ/±ЁѕЇ№¦ДЬЎЈ
ЎЎЎЎ3.2С»·ТєМеІ»ЧгК±Ј¬У¦НЈЦ№№¤ЧчЈ¬ІўѕЯУРМбКѕ/±ЁѕЇ№¦ДЬЎЈ
ЎЎЎЎ3.3МеОВґ«ёРЖчјаІв№¦ДЬТміЈК±Ј¬У¦ѕЯУРМбКѕ/±ЁѕЇ№¦ДЬЎЈ
ЎЎЎЎ3.4їШОВМєІ»У¦ЅцУЙУЪёД±дОВ¶ИЙи¶ЁЦµ¶шЧФ¶ЇФЪЦЖАдєНЦЖИИДЈКЅЦ®јдЗР»»ЎЈ
ЎЎЎЎ4.µзЖш°ІИ«
ЎЎЎЎ4.1ТЅБЖЖчРµІъЖ·ЧўІбЦ¤ґъАн ЙкЗлІъЖ·У¦·ыєПGB 9706.1—2007µДТЄЗуЎЈ
ЎЎЎЎ4.2ѕЯУРЦЖИИ№¦ДЬµДїШОВМєЈ¬У¦·ыєПYY 0834—2011µДТЄЗуЎЈ
ЎЎЎЎ4.3ѕЯУР±ЁѕЇ№¦ДЬµДїШОВМєУ¦·ыєПYY 0709—2009µДТЄЗуЎЈ
ЎЎЎЎ5.µзґЕјжИЭРФ
ЎЎЎЎУ¦·ыєПYY 0505—2012µДТЄЗуЎЈ
ЎЎЎЎ6.»·ѕіКФСй
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІбЦ¤ЙкЗлУ¦·ыєПGB/T 14710—2009µДТЄЗуЎЈ
ЎЎЎЎ7.Нв№Ы
ЎЎЎЎ7.1Цч»ъНв№ЫХыЅаЎўЖбД¤Й«ФуѕщФИЈ¬ОЮЙЛ»®µИИ±ПЭЎЈ
ЎЎЎЎ7.2МєЧУ±нГжУ¦ѕщФИЈ¬ОЮ±дЙ«ЎўНСЙ«ЎўЙшВ©єНїЄБСПЦПуЎЈ
ЎЎЎЎ(ѕЕ)Н¬Т»ЧўІбµҐФЄДЪЧўІбјмСйґъ±нІъЖ·И·¶ЁФФтєНКµАэ
ЎЎЎЎТЅУГїШОВМєН¬Т»ЧўІбµҐФЄДЪЛщјмІвµДІъЖ·Ј¬У¦µ±КЗДЬ№»ґъ±н±ѕЧўІбµҐФЄДЪЖдЛыІъЖ·°ІИ«РФєНУРР§РФµДµдРНІъЖ·ЎЈЅЁТйТФ№¦ДЬЧо¶аЈ¬ДЬёІёЗЧўІбµҐФЄИ«Ії№¦ДЬµДТ»ёц»т¶аёцРНєЕЧчОЄµдРНРНєЕЎЈ
ЎЎЎЎ(К®)ІъЖ·ЙъІъЦЖФмПа№ШТЄЗу
ЎЎЎЎУ¦µ±ГчИ·ТЅУГїШОВМєІъЖ·µДЙъІъ№¤ТХ№эіМЈ¬їЙІЙУГБчіМНјµДРОКЅЈ¬ІўЛµГчЖд№эіМїШЦЖµгЎЈУР¶аёцСРЦЖЎўЙъІъіЎµШЈ¬У¦µ±ёЕКцГїёцСРЦЖЎўЙъІъіЎµШµДКµјКЗйїцЎЈ
ЎЎЎЎ(К®Т»)ІъЖ·µДБЩґІЖАјЫПё»ЇТЄЗу
ЎЎЎЎТЅБЖЖчРµЧўІбЦ¤ґъАнТЅУГїШОВМєІъЖ·µДБЩґІЖАјЫУ¦·ыєПЎ¶ТЅБЖЖчРµЧўІб№ЬАн°м·ЁЎ·(№ъјТКіЖ·Т©Ж·ја¶Ѕ№ЬАнЧЬѕЦБоµЪ4єЕ)єНЎ¶ТЅБЖЖчРµБЩґІЖАјЫјјКхЦёµјФФтЎ·(№ъјТКіЖ·Т©Ж·ја¶Ѕ№ЬАнЧЬѕЦНЁёж2015ДкµЪ14єЕ)µДТЄЗуЎЈ
ЎЎЎЎІОХХЎ¶ГвУЪЅшРРБЩґІКФСйµДµЪ¶юАаТЅБЖЖчРµДїВјЎ·(№ъјТКіЖ·Т©Ж·ја¶Ѕ№ЬАнЧЬѕЦНЁёж2014ДкµЪ12єЕ)(ТФПВјтіЖЎ¶ДїВјЎ·)Ј¬ТЅУГїШОВМєІъЖ·µДБЩґІЖАјЫЧКБПТЄЗуИзПВЈє
ЎЎЎЎ1.МбЅ»Йк±ЁІъЖ·Па№ШРЕПўУлЎ¶ДїВјЎ·ЛщКцДЪИЭµД±И¶ФЧКБПЈє
ЎЎЎЎТЅБЖЖчРµЧўІбЦ¤ґъ°мЙк±ЁІъЖ·Па№ШРЕПўУлЎ¶ДїВјЎ·ЛщКцДЪИЭ(ІъЖ·ГыіЖЎўЅб№№ЧйіЙЎўККУГ±кЧјЎўФ¤ЖЪУГНѕµИ)µД±И¶ФЧКБП;
ЎЎЎЎ2.МбЅ»Йк±ЁІъЖ·УлЎ¶ДїВјЎ·ЦРѕіДЪТСЙПКРН¬Ж·ЦЦТЅБЖЖчРµµД±И¶ФЛµГчЈ¬±И¶ФЛµГчУ¦µ±°ьАЁЎ¶Йк±ЁІъЖ·УлДїВјДЪѕіДЪТСЙПКРН¬Ж·ЦЦТЅБЖЖчРµ±И¶Ф±нЎ·єНПаУ¦Ц§іЦРФЧКБПЎЈ¶Ф±ИДЪИЭУ¦°ьАЁІъЖ·ГыіЖЎў»щ±ѕФАнЎў»ъ№№ЧйіЙЎўРФДЬТЄЗуЎўККУГ·¶О§ЎўК№УГ·Ѕ·ЁµИ;Ц§іЦРФЧКБПКЗЦёЙк±ЁІъЖ·УлЎ¶ДїВјЎ·ІъЖ·µДІоТмРФ¶ФЙк±ЁІъЖ·µД°ІИ«УРР§РФІ»ІъЙъУ°ПмµДАнУЙєНТАѕЭЈ¬їЙТФёЅјюµДРОКЅМṩЎЈ
ЎЎЎЎМбЅ»µДЙПКцЧКБПУ¦ДЬЦ¤ГчЙк±ЁІъЖ·УлЎ¶ДїВјЎ·ЛщКцµДІъЖ·ѕЯУРµИН¬РФЎЈ
ЎЎЎЎ(К®¶ю)ІъЖ·µДІ»БјКВјюАъК·јЗВј
ЎЎЎЎТЅБЖЖчРµЧўІбЙк±ЁёщѕЭ±±ѕ©КРТ©Ж·І»Бј·ґУ¦ЦРРДМṩµДРЕПўЈ¬2008Дк1ФВ15ИХїШОВМєУГУЪ“ёОСЧєуёОУІ»ЇГЕѕІВцёЯС№Цў”»јХЯФЪИ«ВйПВРР“ЖўЗРіэКхјУГЕЖжѕІВц¶ПБчКх”ЦРОЄ»јХЯЙэОВ№эіМЦРФш·ўЙъИзПВІ»БјКВјюЈє»јХЯ±іІїј°УТЦвІїЖ¤·фємЦЧІї·ЦЛ®ЕЭЈ¬Гж»э3%Ј¬Хп¶ПОЄЖ¤·фММЙЛ(ўт¶И)ЎЈ
ЎЎЎЎ·ЦОцФТтЈє
ЎЎЎЎКЦКх№эіМЦРЈ¬їШОВМєКЬЗїµзґЕёЙИЕєуїШОВПµНіК§їШЛщЦВЈ¬їШОВМєИ±·¦ї№µзґЕёЙИЕДЬБ¦Ј¬УЦГ»УРМṩЗРКµїЙРРµДї№ёЙИЕґлК©Ј¬І»БјКВјю·ўЙъУлїШОВМєФЪї№µзґЕёЙИЕ·ЅГжµДЙијЖИ±ПЭЦ±ЅУПа№ШЎЈ
ЎЎЎЎХыёД·Ѕ·ЁЈє
ЎЎЎЎ1.ІОХХYY 0505—2012µзґЕјжИЭТЄЗуєНКФСй±кЧјЈ¬МбёЯїШОВМєІъЖ·ї№µзґЕёЙИЕДЬБ¦ЎЈ
ЎЎЎЎ2.ЙъІъЖуТµРЮёДЛµГчКйЈ¬ПЮЦЖїШОВМєК№УГ·¶О§Ј¬МṩЖдК№УГ»·ѕіµзґЕЗї¶ИµДѕЯМеТЄЗу;ГчИ·№ШјьІїјюЈ¬ИзЈєОВ¶Иґ«ёРЖчЎўОВ¶ИПФКѕЖчµИјаКУєНО¬»¤µД·Ѕ·ЁЈ¬ТФ±гУЪК№УГХЯІЩЧчЎЈ
ЎЎЎЎ3.ТЅБЖ»ъ№№НкЙЖТЅУГЙи±ёК№УГЦЖ¶ИЈ¬°ґХХЛµГчКйТЄЗу¶ФїШОВМєЅшРР¶ЁЖЪО¬»¤єН±ЈСшЈ¬ІўЧцєГПаУ¦јЗВјЎЈФЪК№УГ№эіМЦРУ¦ѕЎБї±ЬГвУлЗїµзґЕёЙИЕЙи±ёН¬К±К№УГЈ¬іцПЦТміЈЗйїцУ¦ЧўТв№ЫІм»јХЯЧґїцЎЈ
ЎЎЎЎ(К®Иэ)СРѕїТЄЗу
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІбёщѕЭЛщЙк±ЁµДІъЖ·Ј¬МṩККУГµДСРѕїЧКБПЎЈ
ЎЎЎЎ1.ІъЖ·РФДЬСРѕї
ЎЎЎЎУ¦µ±МṩІъЖ·РФДЬСРѕїЧКБПТФј°ІъЖ·јјКхТЄЗуµДСРѕїєН±аЦЖЛµГчЈ¬°ьАЁ№¦ДЬРФЎў°ІИ«РФЦё±к(ИзµзЖш°ІИ«УлµзґЕјжИЭЎў·шЙд°ІИ«)ТФј°УлЦКБїїШЦЖПа№ШµДЖдЛыЦё±кµДИ·¶ЁТАѕЭЈ¬ЛщІЙУГµД±кЧј»т·Ѕ·ЁЎўІЙУГµДФТтј°АнВЫ»щґЎЎЈ
ЎЎЎЎ2.ЙъОпПаИЭРФЖАјЫСРѕї
ЎЎЎЎУ¦¶ФіЙЖ·ЦРУл»јХЯєНК№УГХЯЦ±ЅУ»тјдЅУЅУґҐµДІДБПµДЙъОпПаИЭРФЅшРРЖАјЫЎЈ
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІбЦ¤°мАнЙъОпПаИЭРФЖАјЫСРѕїЧКБПУ¦µ±°ьАЁЈєЙъОпПаИЭРФЖАјЫµДТАѕЭєН·Ѕ·Ё;ІъЖ·ЛщУГІДБПµДГиКцј°УлИЛМеЅУґҐµДРФЦК;КµК©»т»нГвЙъОпС§КФСйµДАнУЙєНВЫЦ¤;¶ФУЪПЦУРКэѕЭ»тКФСйЅб№ыµДЖАјЫЎЈ
ЎЎЎЎ3.ГрѕъєНПы¶ѕ№¤ТХСРѕї
ЎЎЎЎЦХ¶ЛУГ»§Пы¶ѕЈєИзККУГЈ¬У¦ГчИ·НЖјцµДПы¶ѕ№¤ТХ(·Ѕ·ЁєНІОКэ)ТФј°ЛщНЖјцПы¶ѕ·Ѕ·ЁИ·¶ЁµДТАѕЭ
ЎЎЎЎ4.ІъЖ·УРР§ЖЪєН°ьЧ°СРѕї
ЎЎЎЎУРР§ЖЪµДИ·¶ЁЈєИзККУГЈ¬У¦µ±МṩІъЖ·УРР§ЖЪµДСйЦ¤±ЁёжЎЈ
ЎЎЎЎ¶ФУЪУРПЮґОЦШёґК№УГµДТЅБЖЖчРµЈ¬У¦µ±МṩʹУГґОКэСйЦ¤ЧКБПЎЈ
ЎЎЎЎ°ьЧ°ј°°ьЧ°НкХыРФЈєФЪРыіЖµДУРР§ЖЪДЪТФј°ФЛКдґўґжМхјюПВЈ¬±ЈіЦ°ьЧ°НкХыРФµДТАѕЭЎЈ
ЎЎЎЎ5.ИнјюСРѕї
ЎЎЎЎТЅБЖЖчРµЧўІбЦ¤°мАнє¬УРИнјюµДІъЖ·Ј¬У¦µ±Мṩһ·ЭµҐ¶АµДТЅБЖЖчРµИнјюГиКцОДµµЈ¬ДЪИЭ°ьАЁ»щ±ѕРЕПўЎўКµПЦ№эіМєНєЛРДЛг·ЁЈ¬ПкѕЎіМ¶ИИЎѕцУЪИнјюµД°ІИ«РФј¶±рєНёґФУіМ¶ИЎЈН¬К±Ј¬У¦іцѕЯ№ШУЪИнјю°ж±ѕГьГы№жФтµДЙщГчЈ¬ІўГчИ·Инјю°ж±ѕµДИ«ІїЧЦ¶Ој°ЧЦ¶Оє¬ТеЈ¬И·¶ЁИнјюµДНкХы°ж±ѕєН·ўРРЛщУГµД±кК¶°ж±ѕЎЈѕЯМеІОјыЎ¶ТЅБЖЖчРµИнјюЧўІбјјКхЙуІйЦёµјФФтЎ·(№ъјТКіЖ·Т©Ж·ја¶Ѕ№ЬАнЧЬѕЦНЁёж2015ДкµЪ50єЕ)µДПа№ШТЄЗуЎЈ
ЎЎЎЎ6.ЖдЛыЧКБП
ЎЎЎЎЦ¤ГчІъЖ·°ІИ«РФЎўУРР§РФµДЖдЛыСРѕїЧКБПЎЈ
ЎЎЎЎ(К®ЛД)ТЅБЖЖчРµІъЖ·ЧўІбЦ¤ґъАнІъЖ·ЛµГчКйєН±кЗ©ТЄЗу
ЎЎЎЎ1.НЁУГТЄЗу
ЎЎЎЎІъЖ·µД±кЦѕЎў±кЗ©єНК№УГЛµГчКйУ¦·ыєПЎ¶ТЅБЖЖчРµЛµГчКйєН±кЗ©№ЬАн№ж¶ЁЎ·(№ъјТКіЖ·Т©Ж·ја¶Ѕ№ЬАнЧЬѕЦБоµЪ6єЕ)µД№ж¶ЁЎЈ
ЎЎЎЎ2.К№УГЛµГчКй
ЎЎЎЎ2.1ІъЖ·ГыіЖЎўРНєЕЎў№жёсЎЈ
ЎЎЎЎ2.2ЧўІбИЛµДГыіЖЎўЧЎЛщЎўБЄПµ·ЅКЅј°КЫєу·юОсµҐО»Ј¬ЅшїЪТЅБЖЖчРµ»№У¦µ±ФШГчґъАнИЛµДГыіЖЎўЧЎЛщј°БЄПµ·ЅКЅЎЈ
ЎЎЎЎ2.3ЙъІъЖуТµµДГыіЖЎўЧЎЛщЎўЙъІъµШЦ·ЎўБЄПµ·ЅКЅј°ЙъІъРнїЙЦ¤±аєЕЈ¬ОЇНРЙъІъµД»№У¦µ±±кЧўКЬНРЖуТµµДГыіЖЎўЧЎЛщЎўЙъІъµШЦ·ЎўЙъІъРнїЙЦ¤±аєЕЎЈ
ЎЎЎЎ2.4ТЅБЖЖчРµІъЖ·ЧўІбЦ¤±аєЕЎЈ
ЎЎЎЎ2.5ІъЖ·јјКхТЄЗуµД±аєЕЎЈ
ЎЎЎЎ2.6ІъЖ·РФДЬЎўЦчТЄЅб№№ЧйіЙЎўККУГ·¶О§ЎЈ
ЎЎЎЎ2.7ЅыјЙЦўЎўЧўТвКВПоТФј°ЖдЛыРиТЄѕЇКѕ»тХЯМбКѕµДДЪИЭЎЈ
ЎЎЎЎ2.8°ІЧ°єНК№УГЛµГч»тХЯНјКѕЎЈ
ЎЎЎЎ2.9ІъЖ·О¬»¤єН±ЈСш·Ѕ·ЁЈ¬МШКвґўґжЎўФЛКдМхјюЎў·Ѕ·ЁЎЈ
ЎЎЎЎ2.10ЙъІъИХЖЪЈ¬К№УГЖЪПЮ»тХЯК§Р§ИХЖЪЎЈ
ЎЎЎЎ2.11ЕдјюЗ嵥Ј¬°ьАЁЕдјюЎўёЅКфЖ·ЎўЛрєДЖ·ёь»»ЦЬЖЪТФј°ёь»»·Ѕ·ЁµДЛµГчµИЎЈ
ЎЎЎЎ2.12ТЅБЖЖчРµ±кЗ©ЛщУГµДНјРОЎў·ыєЕЎўЛхРґµИДЪИЭµДЅвКНЎЈ
ЎЎЎЎ2.13ЛµГчКйµД±аЦЖ»тХЯРЮ¶©ИХЖЪЎЈ
ЎЎЎЎ2.14АдДэЖч¶ЁЖЪО¬»¤»тЗеЅаµД·Ѕ·ЁєНЖµґОЎЈ
ЎЎЎЎ2.15їШОВМєОВ¶ИґпµЅОИМ¬КдіцЛщРиµДК±јдЎЈ
ЎЎЎЎ2.16·АЦ№С»·ТєМеЅб±щµДЛµГч(ИзУР)ЎЈ
ЎЎЎЎ2.17јмІйЎўІ№ід»тёь»»С»·ТєМеµД·Ѕ·ЁєНЖµґОЎЈ
ЎЎЎЎЧўЈєУ¦ГчИ·“І№ідЎўёь»»С»·ТєМеУ¦ФЪНЈ»ъЧґМ¬ПВНкіЙ”ЎЈ
ЎЎЎЎ2.18ОВ¶Иґ«ёРЖчµДРЈЧјЦЬЖЪј°ЧФРЈ·Ѕ·ЁЎЈ
ЎЎЎЎ2.19У¦±кГчМеОВґ«ёРЖчІ»їЙµҐ¶АЧчОЄМеОВјаІвЧ°ЦГК№УГЎЈ
ЎЎЎЎ2.20У¦µ±ФЪЛµГчКйЦР±кГчµДЖдЛыДЪИЭ(ИзУР)ЎЈ
ЎЎЎЎ3.±кЗ©
ЎЎЎЎ3.1ІъЖ·ГыіЖЎўРНєЕЎў№жёсЎЈ
ЎЎЎЎ3.2ЧўІбИЛµДГыіЖЎўЧЎЛщЎўБЄПµ·ЅКЅЈ¬ЅшїЪТЅБЖЖчРµ»№У¦µ±ФШГчґъАнИЛµДГыіЖЎўЧЎЛщј°БЄПµ·ЅКЅЎЈ
ЎЎЎЎ3.3ТЅБЖЖчРµЧўІбЦ¤±аєЕЎЈ
ЎЎЎЎ3.4ЙъІъЖуТµµДГыіЖЎўЧЎЛщЎўЙъІъµШЦ·ЎўБЄПµ·ЅКЅј°ЙъІъРнїЙЦ¤±аєЕЈ¬ОЇНРЙъІъµД»№У¦µ±±кЧўКЬНРЖуТµµДГыіЖЎўЧЎЛщЎўЙъІъµШЦ·ЎўЙъІъРнїЙЦ¤±аєЕЎЈ
ЎЎЎЎ3.5ЙъІъИХЖЪЈ¬К№УГЖЪПЮ»тХЯК§Р§ИХЖЪЎЈ
ЎЎЎЎ3.6µзФґБ¬ЅУМхјюЎўКдИ빦ВКЎЈ
ЎЎЎЎ3.7ТАѕЭІъЖ·МШРФУ¦µ±±кЧўµДНјРОЎў·ыєЕТФј°ЖдЛыПа№ШДЪИЭЎЈ
ЎЎЎЎ3.8±ШТЄµДѕЇКѕЎўЧўТвКВПоЎЈ
ЎЎЎЎ3.9МШКвґўґжЎўІЩЧчМхјю»тХЯЛµГчЎЈ
ЎЎЎЎ3.10К№УГЦР¶Ф»·ѕіУРЖЖ»µ»тХЯёєГжУ°ПмµДТЅБЖЖчРµЈ¬Жд±кЗ©У¦µ±°ьє¬ѕЇКѕ±кЦѕ»тХЯЦРОДѕЇКѕЛµГчЎЈ
ЎЎЎЎТЅБЖЖчРµЧўІбЦ¤ґъАн±кЗ©ТтО»ЦГ»тХЯґуРЎКЬПЮ¶шОЮ·ЁИ«Ії±кГчЙПКцДЪИЭµДЈ¬ЦБЙЩУ¦µ±±кЧўІъЖ·ГыіЖЎўРНєЕЎў№жёсЎўЙъІъИХЖЪєНК№УГЖЪПЮ»тХЯК§Р§ИХЖЪЈ¬ІўФЪ±кЗ©ЦРГчИ·“ЖдЛыДЪИЭПкјыЛµГчКй”ЎЈ
ЙоЫЪєиФ¶ТЅБЖЖчРµЧЙСЇУРПЮ№«ЛѕКЗТ»јТјјКхЧЁТµµДТЅБЖЖчРµЧЙСЇ·юОс№«ЛѕЈ¬ЧЁЧўМṩȫ№ъёчµШИзЈєЙоЫЪЎў№гЦЭЎў¶«ЭёЎўЦРЙЅЎў·рЙЅЎўі±ЦЭЎўЛіµВЎўЙПєЈЎўОч°ІЎўЦШЗмЎўіЙ¶јµИЦЄГыіЗКРµДТЅБЖЖчРµБмУтјјКхЧЙСЇ·юОсЎЈЧЁТµµДТЅБЖЖчРµЧўІбЧЙСЇґъ°мАнЎўТЅБЖЖчРµІъЖ··ЦАаЅз¶Ёґъ°мАнЎўТЅБЖЖчРµЙъІъРнїЙЦ¤ЎўИэАаТЅБЖЖчРµѕУЄРнїЙЦ¤Ўў¶юАаТЅБЖЖчРµѕУЄ±ё°ёЎўЅшїЪТЅБЖЖчРµЧўІбЎўТ»АаТЅБЖЖчРµІъЖ·±ё°ёј°ЙъІъ±ё°ёЎўFDAЧўІбЎўISO13485ИПЦ¤Ўў CEИПЦ¤ЎўјЖБїЖчѕЯЙъІъРнїЙЦ¤ЎўБЩґІКФСйЎўіцїЪЦ¤Ј¬ЧФУЙПъКЫЦ¤µИґъ°мАнЎўТЅБЖЖчРµЦКБї№ЬАнМеПµИПЦ¤ОДјюµДЅЁБўј°МеПµУл№эіМИ·ИПОДјюµДЅЁБў (ИзЈєISO9001Ўў ISO13485 ЎўGMPЎў CEЎўQSR820ЎўCMDCAS)ІъЖ·јмІвЈ¬БЩґІКФСйј°ГвБЩґІЧКБП±аРґЎўІъЖ·јјКхТЄЗуЦЖ¶©ЎўјјКхОДјю±аРґёЁµјЎўµзґЕјжИЭХыёДЎўТЅБЖЖчРµ№гёжЕъОДЙкЗл°мАнЎўЅаѕ»КТЅЁЙиЦёµјµИ·юОсЈ¬јјКхЧЁТµЈ¬іПРЕ·юОсЈ¬ґъАн·СУГµНЈ¬»¶УДъЧЙСЇЈЎ
ЎЎЎЎ
|

