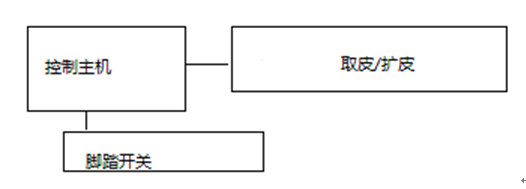
|
ЁЁЁЁ
ЁЁ вНСЦЦїаЕзЂВсОнжиЧьЪавЉЦЗММЪѕЩѓЦРШЯжЄжааФ2020Фъ10дТ19ШеЙигкЗЂВМЁЖвНСЦЦїаЕЕчДХМцШнадММЪѕЦРМлжИФЯЁЗЕФЭЈжЊЃЌЮЊЗўЮёЮвЪавНСЦЦїаЕаавЕЕчДХМцШнАВШЋЦРМлЙЄзїЃЌгааЇПижЦЕкЖўРргадДвНСЦЦїаЕВњЦЗжЪСПАВШЋЗчЯеЃЌВЂНјвЛВНЬсЩ§вНСЦЦїаЕВњЦЗПЦбЇДДаТМрЙмФмСІЃЌжиЧьЪавЉЦЗММЪѕЩѓЦРШЯжЄжааФзщжЏБржЦСЫЁЖвНСЦЦїаЕЕчДХМцШнадММЪѕЦРМлжИФЯЁЗЁЃ
ЁЁЁЁБОжИФЯвРОнЁЖЙигкYY 0505-2012вНСЦЦїаЕаавЕБъзМЪЕЪЉгаЙиЙЄзївЊЧѓЕФЭЈжЊЁЗ(ЪГвЉМрАьаЕЁВ2012ЁГ151КХ)ЁЂYY 0505-2012ЁЖвНгУЕчЦјЩшБИ Ек1-2ВПЗжЃКАВШЋЭЈгУвЊЧѓ ВЂСаБъзМЃКЕчДХМцШн вЊЧѓКЭЪдбщЁЗЕШЯрЙиЗЈЙцБъзМЮФМўБржЦЃЌАќРЈе§ЮФЁЂИНТМКЭБржЦЫЕУїШ§ИіВПЗжФкШнЁЃе§ЮФКЌ9ИіеТНкЃЌжївЊа№ЪіЕчДХМцШнЛљБОддђЁЂММЪѕПМСПЁЂЕчДХМцШнадУшЪіЮФЕЕвЊЧѓЁЂвНСЦЦїаЕВњЦЗзЂВсЩъБЈзЪСЯвЊЧѓЁЂЩѓВщЙизЂЕуЕШФкШн;ИНТМЙВ6ИіЃЌИНТМ1жСИНТМ5зїЮЊЖдзЂВсМьбщЙ§ГЬЁЂВњЦЗММЪѕвЊЧѓЁЂЫЕУїЪщБраДвдМАЖдзЂВсЩъЧыШЫЫЭМьзЪСЯЬюаДЕФВЮПМжИв§ЃЌИНТМ6зїЮЊЖдЯжааЕчДХМцШнадзЈгУБъзМЕФЪОР§ЁЃ
ЁЁЁЁБОжИФЯПЩвдЮЊжиЧьЪавНСЦЦїаЕЩњВњЦѓвЕВњЦЗЩшМЦПЊЗЂЬсЙЉЕчДХМцШнадЗНУцЕФММЪѕжИв§ВЮПМЃЌЮЊЯрЙижЪСПЦРМлВПУХКЭШЫдБЬсЙЉдкЗЈЙцПђМмЯТЕФЕчДХМцШнадММЪѕЦРМлЗНЗЈВЮПМЁЃБОжИФЯВЛЪєгкЗЈЙцЮФМўЧПжЦжДааЃЌПЩзїЮЊзЂВсЩъБЈЁЂзЂВсМьбщЁЂММЪѕЩѓЦРЕШЗНУцЕФВЮПМЙЄОпКЭЙЄзїжИв§ЁЃБОжИФЯИљОнЙњМвЯрЙиЗЈЙцКЭБъзМБфЛЏНјааЭъЩЦИќаТЁЃ
ЁЁ
ЁЁЁЁБОжИФЯжМдкЮЊжиЧьЪавНСЦЦїаЕЩъБЈЦѓвЕзЂВсЩъБЈЬсЙЉЕчДХМцШнадЗНУцЕФММЪѕжИЕМЃЌЭЌЪБЙцЗЖвНСЦЦїаЕЕчДХМцШнадЕФММЪѕЦРМлвЊЧѓЃЌЭГвЛЦРМлГпЖШЁЃ
ЁЁЁЁБОжИФЯЪЧЖджиЧьЪаЕкЖўРрвНСЦЦїаЕЕчДХМцШнадЕФвЛАуадвЊЧѓЃЌжЦдьЩЬгІИљОнвНСЦЦїаЕЕФРрБ№КЭЬиаджЦЖЈММЪѕЮФМўЃЌХаЖЯжИФЯжаЕФОпЬхФкШнЪЧЗёЪЪгУЃЌВЛЪЪгУФкШнЯъЪіРэгЩЁЃжЦдьЩЬвВПЩВЩгУЦфЫћТњзуЗЈЙцКЭБъзМвЊЧѓЕФЬцДњЗНЗЈЃЌЕЋгІЬсЙЉЯъОЁЕФбаОПзЪСЯТлЪіЦфПЩааадЁЃ
ЁЁЁЁБОжИФЯЪЧдкЯжааЗЈЙцКЭБъзМЬхЯЕвдМАЕБЧАШЯжЊЫЎЦНЯТЁЂВЂВЮПМСЫЙњЭтЗЈЙцгыжИФЯЁЂЙњМЪБъзМгыММЪѕБЈИцжЦЖЈЕФЁЃЫцзХЗЈЙцКЭБъзМЕФВЛЖЯЭъЩЦЃЌвдМАШЯжЊЫЎЦНКЭММЪѕФмСІЕФВЛЖЯЬсИпЃЌЯрЙиФкШнвВНЋЪЪЪБНјаааоЖЉЁЃ
ЁЁЁЁБОжИФЯЪЧЖджЦдьЩЬКЭЩѓВщШЫдБЕФжИЕМадЮФМўЃЌПЩзїЮЊвНСЦЦїаЕзЂВсЩъБЈЁЂзЂВсМьбщЁЂММЪѕЩѓЦРЕШЗНУцЕФВЮПМЙЄОпКЭЙЄзїжИв§ЃЌВЛАќРЈЩѓЦРЩѓХњЫљЩцМАЕФааеўЪТЯюЃЌврВЛзїЮЊЗЈЙцЧПжЦжДааЃЌгІдкзёбЯрЙиЗЈЙцЕФЧАЬсЯТЪЙгУБОжИФЯЁЃ
ЁЁЁЁБОжИФЯеыЖдвНСЦЦїаЕЕчДХМцШнадЕФЬиЪтадЃЌдкЯжааЗЈЙцвЊЧѓЯТНјвЛВНУїШЗСЫЖдвНСЦЦїаЕЕчДХМцШнадЕФвЊЧѓЃЌЬиБ№ЪЧЖдвНСЦЦїаЕЕчДХМцШнадВтЪдЕФЙЄзїФЃЪНбЁШЁЁЂПЙШХЖШВПЗжЛљБОадФмКЭМьВтЗНЗЈЕФжЦЖЈвЊЧѓЁЂПЙШХЖШЗћКЯадХаОнвЊЧѓзїГіЯъЯИЙцЖЈЁЃБОжИФЯЪЧвНСЦЦїаЕЕчДХМцШнадЕФЭЈгУММЪѕЦРМлжИФЯЃЌЦфЫћЩцМАвНСЦЦїаЕВњЦЗЕФжИФЯПЩВЮееБОжИФЯНјаагаеыЖдадЕФЕїећЁЂаоИФКЭЭъЩЦЁЃ
ЁЁЁЁвЛЁЂЗЖЮЇ
ЁЁЁЁБОжИФЯЪЪгУгкжиЧьЪавНСЦЦїаЕЕчДХМцШнадЕФММЪѕЦРМлЃЌЪЪгУЪаФкЕкЖўРрвНСЦЦїаЕзЂВсМьбщЁЂММЪѕЩѓЦРЁЃЕквЛРрвНСЦЦїаЕБИАИЫљЩцМАФкШнПЩВЮеежДааЁЃ
ЁЁЁЁЖўЁЂЛљБОддђ
ЁЁЁЁ(вЛ)ЛљБОИХПі
ЁЁЁЁвНСЦЦїаЕЕФЕчДХМцШнадЪЧжИЩшБИЛђЯЕЭГдкЦфЕчДХЛЗОГжаФме§ГЃЙЄзїЧвВЛЖдЛЗОГжаШЮКЮЪТЮяЙЙГЩВЛФмГаЪмЕФЕчДХЩЇШХЕФФмСІЁЃ
ЁЁЁЁвНСЦЦїаЕЕчДХМцШнадАќРЈЗЂЩфВПЗжКЭПЙШХЖШВПЗжЁЃЗЂЩфВПЗжАќРЈЮоЯпЕчвЕЮёЕФБЃЛЄКЭЙЋЙВЕчЭјЕФБЃЛЄЃЌЦфжаЮоЯпЕчвЕЮёЕФБЃЛЄАќРЈДЋЕМЗЂЩфКЭЗјЩфЗЂЩфЃЌЙЋЙВЕчЭјЕФБЃЛЄАќРЈаГВЈЪЇецЁЂЕчбЙВЈЖЏКЭЩСЫИ;ПЙШХЖШВПЗжАќРЈОВЕчЗХЕчПЙШХЖШЁЂЩфЦЕЕчДХГЁЗјЩфПЙШХЖШЁЂЕчПьЫйЫВБфТіГхШКПЙШХЖШЁЂРЫгППЙШХЖШЁЂЩфЦЕИагІЕФДЋЕМЩЇШХПЙШХЖШЁЂЙЄЦЕДХГЁПЙШХЖШМАЕчбЙднНЕЁЂЖЬЪБжаЖЯКЭЕчбЙБфЛЏПЙШХЖШЁЃИїВПЗжФкШнОљгаЖдгІЕФЪЭвхКЭБъзМвЊЧѓЃЌвдYY 0505ЮЊР§ЃЌЪОвтШчЯТЃК
ЁЁЁЁЭМ1 ЕчДХМцШнадБъзМЖдгІЙиЯЕЭМ
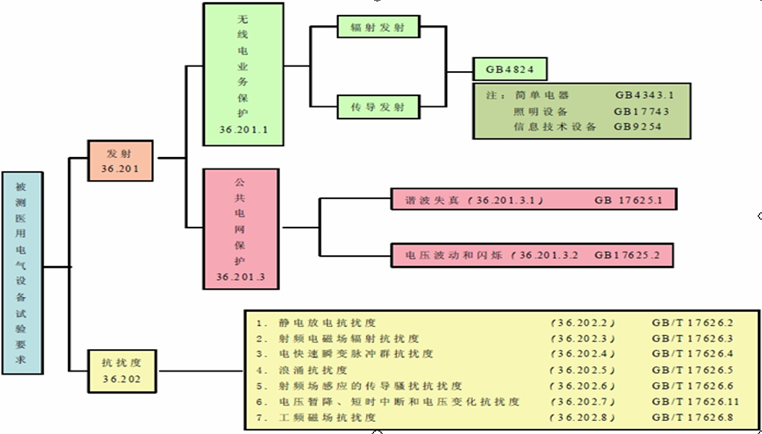
ЁЁЁЁвЊДяЕНЕчДХМцШнадЕФФПЕФЃЌвЛЗНУцвЊПижЦЩшБИЖдЦфЫћЩшБИЕФЕчДХИЩШХЃЌСэвЛЗНУцвЊПижЦЩшБИздЩэЕФЕчДХУєИаЖШЃЌМДЬсИпЩшБИздЩэЕФЕчДХПЙИЩШХФмСІЁЃЕчДХИЩШХвВГЦЮЊЕчДХЩЇШХЃЌЦфРДдДПЩЗжЮЊСНДѓРрЃКздШЛЩЇШХдДКЭШЫЮЊЩЇШХдДЁЃЦфжаЃЌздШЛЩЇШХдДЪЧжИАќРЈЕиЧђЩЯИїДІРзЕчВњЩњЕФЬьЕчдыЩљЃЌЬЋбєКкзгБЌеЈКЭЛюЖЏВњЩњЕФдыЩљвдМАвјКгЯЕЕФгюжцдыЩљ;ШЫЮЊЩЇШХдДЪЧжИЕчЦїЛђЦфЫћгУЕчзАжУВњЩњЕФЕчДХЩЇШХЃЌШчЃКЙЄвЕЁЂПЦбЇКЭвНСЦЩфЦЕЩшБИ(ISM)ЫљВњЩњЕФЕчДХЩЇШХЁЃЕчЦјЩшБИжЎМфжївЊЭЈЙ§ЕМЬхДЋЕМЁЂПеМфЗјЩфКЭЯрСкЯпТЗМфёюКЯЭООЖДЋВЅЁЃгааэЖрЕчЦїЕчзгВњЦЗЖдЕчДХИЩШХЗЧГЃУєИаЃЌГЦЮЊЕчДХУєИаЩшБИЁЃЕчДХИЩШХдДЁЂДЋВЅЭООЖКЭЕчДХУєИаЩшБИМДЙЙГЩСЫЕчДХИЩШХЕФШ§вЊЫиЁЃ
ЁЁЁЁЕчДХИЩШХЖдвНСЦЦїаЕдьГЩЕФКѓЙћЭљЭљЪЧЗЧГЃбЯжиЕФЁЃЙњФкЭтгаЙиЕчДХИЩШХв§Ц№вНСЦЪТЙЪЕФБЈЕРТХМћВЛЯЪЃЌИљОнЙњЭтШЈЭўЛњЙЙЕФБЈИцЃЌзд1973ФъжС1993ФъЕФ20ФъМфЃЌдјЪеЕНвЩЮЊвђвНСЦЦїаЕЪмЕчДХИЩШХв§ЗЂЕФЪТЙЪБЈИцГЌЙ§100МўЃЌЦфжаБЛШЯЖЈЮЊЕчДХИЩШХв§Ц№ЕФЪТЙЪдМеМ10%ЁЃИљОнЙњЭтШЈЭўЛњЙЙЗЂВМЕФСэЭтвЛЗнбаОПБЈИцЯдЪОЃЌДг1994Фъ1дТжС2005Фъ3дТЃЌ550ЗнВЛСМЗДгІЪТМўжаЃЌга73.6%ЪЧгЩПЩвЩЕФЕчДХИЩШХдьГЩЕФЁЃЖјдкетаЉПЩвЩЕФЕчДХИЩШХдьГЩЕФВЛСМЪТМўжаЃЌЫРЭіКЭжТЩЫЕФБШР§ДяЕНСЫ43.5%ЁЃДг1994ФъЕН2005ФъМфЕчДХИЩШХЕФВЛСМЪТМўБЈИцЪ§СПЯдЪОж№ФъЕндіЕФЧїЪЦЁЃЮвЙњвВгаРрЫЦЕФБЈЕРЃЌЗЂЯжЖрЦ№вНСЦЪТЙЪЕФзяП§ЛіЪзОљЮЊЕчДХИЩШХЁЃ
ЁЁЁЁвНСЦЦїаЕзЂВсДњРэЯжДњвНСЦЦїаЕжаЃЌвНгУЕчЦјЩшБИКЭЯЕЭГвдМАЬхЭтеяЖЯЕШЩшБИВЛНіЪЙгУСЫИїжжИпУєИаадЕчзгЦїМўЁЂЕчзгдЊМўЃЌВЂЧвгыЕчФдЁЂвЦЖЏЭЈбЖЯЕЭГЕШНсКЯаЮГЩдЖГЬвНСЦеяЖЯЭјТчЃЌЫќУЧдкЙЄзїЪБЯђжмЮЇЗЂЩфВЛЭЌЦЕТЪЗЖЮЇЁЂВЛЭЌЕчДХГЁЧПЖШЕФгагУЛђЮогУЕФЕчДХВЈЃЌгАЯьЮоЯпЕчЙуВЅЭЈбЖвЕЮёКЭжмЮЇЦфЫћЩшБИЕФЙЄзїЃЌЖјЧвЫќУЧдкЙВЭЌЕФЕчДХЛЗОГжаЛЙПЩФмЪмЕНжмЮЇЕчСІЁЂЕчзгЩшБИвдМАЦфЫћвНСЦЩшБИЕФЕчДХИЩШХЁЃвђДЫЃЌвНСЦЦїаЕЕчДХМцШнадЪЧвНСЦЦїаЕАВШЋадКЭгааЇадЕФживЊзщГЩВПЗжжЎвЛЁЃ
ЁЁЁЁ(Жў)ЛљБОЫМТЗ
ЁЁЁЁВњЦЗЕФЕчДХМцШнадУЛгаЮяРэЪЕЬхЃЌдкПЊЗЂКЭЪЙгУЙ§ГЬжаЛЗОГКЭШЫЮЊвђЫигАЯьЮоДІВЛдкЃЌМгжЎвНСЦЦїаЕЕФжжРрЖрбљадЁЂНсЙЙКЭЙІФмИДдгЃЌЧввђЮЊЕчДХМцШнадВтЪдгЩгкЪБМфКЭГЩБОЕФЯожЦВЛФмЧюОЁЫљгаЧщПіЃЌЫљвддкОпЬхВтЪдЙ§ГЬжаЭљЭљГіЯжДэХаКЭТЉХаЯжЯѓЁЃЭЌЪБЃЌЩшБИЕФФЃЪНбЁдёЁЂВтЪдбљЦЗЕФВМжУЧщПіЁЂзщКЯЪНбљЦЗЕФСЊКЯЪЙгУЁЂЛљБОадФмКЭВтЪдЗНЗЈЕФжЦЖЈвдМАЗћКЯадХаОнЕФШЗЖЈОљЛсжБНггАЯьВњЦЗВтЪдНсЙћЃЌгЩгкЛЙДцдкЪдбщНсЙћИДЯжадНЯВюЕФЮЪЬтЃЌВтЪдНсЙћЭљЭљВЛФмЗДгГВњЦЗЕФецЪЕЕчДХМцШнадЁЃвђДЫЃЌЖдвНСЦЦїаЕЕчДХМцШнадЕФПЭЙлЁЂГфЗжЦРМлЪЧБЃеЯВњЦЗжЪСПЕФживЊвђЫиЁЃ
ЁЁЁЁМјгквНСЦЦїаЕЕчДХМцШнадЕФЬиадЃЌжЛгаЛљгквНСЦЦїаЕЪЪгУЗЖЮЇ/дЄЦкгУЭОЃЌНсКЯЗчЯеЙмРэЁЂжЪСПЬхЯЕЙ§ГЬЙмРэКЭЙњМвБъзМЕФвЊЧѓВХФмБЃжЄЦРМлЕФПЭЙлЁЂГфЗжЁЃ
ЁЁЁЁвНСЦЦїаЕзЂВсзЩбЏЩъЧыШЫгІЕБдквНСЦЦїаЕВњЦЗШЋЩњУќжмЦкЙ§ГЬжаГжајЙизЂЕчДХМцШнадЕФЗћКЯадЮЪЬтЃЌАќРЈвНСЦЦїаЕВњЦЗЕФЩшМЦПЊЗЂЁЂЩњВњЁЂЯњЪлКЭЮЌЛЄЁЃЭЌЪБЃЌзЂВсЩъЧыШЫгІЕБНсКЯздЩэжЪСПЙмРэЬхЯЕЕФвЊЧѓКЭвНСЦЦїаЕВњЦЗЬиЕуРДБЃжЄвНСЦЦїаЕВњЦЗЕФЕчДХМцШнадЗћКЯвЊЧѓЃЌАќРЈЩЯЪаЧАКЭЩЯЪаКѓЕФЯрЙивЊЧѓЃЌШчЗчЯеЙмРэЁЂЩшМЦПЊЗЂЁЂВњЦЗЮЌЛЄМАгУЛЇИцжЊЕШвЊЧѓЁЃ
ЁЁЁЁвНСЦЦїаЕВњЦЗдкЪЙгУЙ§ГЬжаГЃгыЗЧзЂВсЩъЧыШЫдЄЦкЕФЩшБИЛђЯЕЭГЯрСЌНгЃЌетОЭЪЙЕУзЂВсЩъЧыШЫздЩэФбвдПижЦКЭБЃжЄвНСЦЦїаЕВњЦЗЕФЕчДХМцШнадЗћКЯвЊЧѓЁЃвђДЫЃЌвНСЦЦїаЕЕФЕчДХМцШнадашвЊзЂВсЩъЧыШЫЁЂгУЛЇКЭЦфЫћдЄЦкХфКЯЪЙгУЕФЩшБИЛђЯЕЭГЙЉгІЩЬЙВЭЌХЌСІКЭЭЈСІКЯзїВХФмЕУвдБЃеЯЁЃЕЋЪЧетВЂВЛвтЮЖзХзЂВсЩъЧыШЫПЩвдУтГ§ЖдвНСЦЦїаЕЕчДХМцШнадЗћКЯвЊЧѓЕФЯрЙид№ШЮЃЌзЂВсЩъЧыШЫгІЕББЃжЄвНСЦЦїаЕВњЦЗздЩэЕФЕчДХМцШнадЗћКЯвЊЧѓЃЌВЂУїШЗгыЦфдЄЦкЯрСЌЩшБИЛђЯЕЭГЕФвЊЧѓЃЌДгЖјБЃжЄвНСЦЦїаЕВњЦЗЕФАВШЋадКЭгааЇадЁЃ
ЁЁЁЁМјгквНСЦЦїаЕЕчДХМцШнадОпгагАЯьвђЫиЖрЁЂЩцМАУцЙуЁЂзЈвЕадЧПКЭЮЃКІадИпЕШЬиЕуЃЌЕЅЖРПМТЧвНСЦЦїаЕВњЦЗЕФЕчДХМцШнадБъзМЗћКЯадВЛзувдБЃжЄЦфЕчДХМцШнадЦРМлГфЗжЃЌвђДЫЖдгкгывНСЦЦїаЕЕчДХМцШнадгаЙиЕФвНСЦЦїаЕВњЦЗзЂВсЩъБЈзЪСЯЭГвЛНјаавЊЧѓЁЃ
ЁЁЁЁШ§ЁЂЗЂЩфВПЗжЕФММЪѕПМСП
ЁЁЁЁвНСЦЦїаЕЕчДХМцШнадЗЂЩфВПЗжжаЮоЯпЕчвЕЮёЕФБЃЛЄЕФММЪѕПМСПЛљгкGB 4824ЁЖЙЄвЕЁЂПЦбЇКЭвНСЦЩшБИ ЩфЦЕЩЇШХЬиадЯожЕКЭВтСПЗНЗЈЁЗгшвдШЗЖЈЪєадЃЌгУгкХаЖЈвНСЦЦїаЕЮоЯпЕчвЕЮёЕФБЃЛЄЕФЦРМлзщБ№КЭРрБ№ЁЃ
ЁЁЁЁИљОнGB 4824МАжЦдьЩЬЙцЖЈЕФдЄЦкгУЭОЃЌвНСЦЦїаЕЭЈГЃЗжЮЊ1зщКЭ2зщЩшБИЃЌAРрКЭBРрЩшБИЁЃ
ЁЁЁЁ1зщЩшБИЪЧГ§БъзМЗЖЮЇФкЙцЖЈЕФ2зщЩшБИЭтЕФЦфЫћЩшБИЁЃШчЃКГЌЩљеяЖЯ/жЮСЦЩшБИЁЂXЩфЯпеяЖЯ/жЮСЦЩшБИЁЃ
ЁЁЁЁ2зщЩшБИАќРЈвдЕчДХЗјЩфЁЂИаадКЭ/ЛђШнадёюКЯаЮЪНЃЌгавтВњЩњВЂЪЙгУЛђОжВПЪЙгУ9 kHzЁЋ400 GHzЦЕЖЮФкЩфЦЕФмСПЕФЃЌЫљгагУгкВФСЯДІРэЛђМьбщ/ЗжЮіФПЕФЃЌЛђгУгкДЋЪфЕчДХФмСПЕФЙЄПЦвНЩфЦЕЩшБИЁЃШчЃКДХЙВеёГЩЯёЯЕЭГЁЂЮЂВЈжЮСЦЩшБИЁЃ
ЁЁЁЁЩшБИЗжзщОйР§ЯъМћGB 4824ИНТМAКЭYY 0505ИНТМCЁЃ
ЁЁЁЁAРрЩшБИЪЧжИЗЧОгзЁЛЗОГКЭВЛжБНгСЌНгЕНзЁеЌЕЭбЙЙЉЕчЭјЩшЪЉжаЪЙгУЕФЩшБИЁЃ
ЁЁЁЁBРрЩшБИЪЧжИМвгУЩшБИКЭжБНгСЌНгЕНзЁеЌЕЭбЙЙЉЕчЭјЩшЪЉжаЪЙгУЕФЩшБИЁЃ
ЁЁЁЁЭЈГЃЃЌЙЋЙВЕчЭјзїЮЊ“ЬсЙЉИјНЈжўЮягУгкМвЭЅФПЕФЙЋЙВЕЭбЙЙЉЕчЭј”ЁЃЖјгУдкЯёвНдКЕШГЁЫљжаЕФЩшБИКЭЯЕЭГЪЧВЛСЌНгЕНЙЋЙВЕчЭјЕФЃЌетаЉГЁЫљЕФЭјЕчдДСЌНгЪЧЭЈЙ§БфбЙЦїЛђХфЕчеОгыЙЋЙВЕЭбЙЙЉЕчЭјИєРыЕФЁЃ
ЁЁЁЁвНСЦЦїаЕВњЦЗзЂВсДњРэДЫЭтЃЌАДYY 0505вЊЧѓЃЌжЛАќРЈЯёЕчЖЏЛњКЭПЊЙивЛРрМђЕЅЕчЦјЦїМўЃЌвдМАВЛЪЙгУШЮКЮВњЩњЛђЪЙгУ9 kHzвдЩЯЦЕТЪЕФЕчзгЕчТЗ(ШчЃКвЛаЉбРзъЛњЁЂКєЮќЛњКЭЪжЪѕЬЈ)ЕФвНгУЕчЦјЩшБИЃЌПЩвРОнGB 4343.1РДЗжРрЃЌЗжРрНіЯогкЕЅЛњЩшБИЃЌВЛЪЪгУгкЯЕЭГЛђзгЯЕЭГЁЃ
ЁЁЁЁгУгквНСЦгУЭОЕФееУїЩшБИ(ШчЃКXЙтЦЌЕФееУїЩшБИЁЂЪжЪѕЪвЕФееУїзАжУ)ПЩАДGB 17743ЗжРрЃЌЗжРрНіЯогкЕЅЛњЩшБИЃЌВЛЪЪгУгкЯЕЭГЛђзгЯЕЭГЁЃ
ЁЁЁЁгыЩшБИЛђЯЕЭГСЌНгЕФаХЯЂММЪѕЩшБИПЩАДGB 9254ЗжРрЃЌЕЋGB 9254ЕФBРрЩшБИПЩгыGB 4824ЕФAРрЛђBРрЯЕЭГвЛЦ№ЪЙгУЃЌGB 9254ЕФAРрЩшБИНіПЩгыGB 4824ЕФAРрЯЕЭГвЛЦ№ЪЙгУЁЃ
ЁЁЁЁГ§ЛљЦЕЕФЕкШ§ДЮаГВЈЭтЃЌЦфЫћЦЕТЪФмТњзуGB 4824 2зщBРрЕчДХЗјЩфЩЇШХЯожЕЕФзЈгУЩшБИЛђЯЕЭГЃЌЭЌЪБЛљЦЕЕФЕкШ§ДЮаГВЈВЛФмТњзуGB 4824 2зщBРрЕчДХЗјЩфЩЇШХЯожЕЕФвЊЧѓЃЌЕЋТњзу2зщAРрЕчДХЗјЩфЩЇШХЯожЕЕФЩшБИЛђЯЕЭГПЩЗжЮЊAаЭзЈгУЩшБИКЭЯЕЭГЁЃ
ЁЁЁЁвНСЦЦїаЕЗЂЩфВПЗжжаЙЋЙВЕчЭјЕФБЃЛЄЕФММЪѕПМСПЛљгкGB 17625.1ЁЖЕчДХМцШн ЯожЕ аГВЈЕчСїЗЂЩфЯожЕ(ЩшБИУПЯрЪфШыЕчСї≤16A)ЁЗКЭGB 17625.2ЁЖЕчДХМцШн ЯожЕ ЖдУПЯрЖюЖЈЕчСї16AЧвЮоЬѕМўНгШыЕФЩшБИдкЙЋЙВЕЭбЙЙЉЕчЯЕЭГжаВњЩњЕФЕчбЙБфЛЏЁЂЕчбЙВЈЖЏКЭЩСЫИЕФЯожЦЁЗгшвдШЗЖЈЪєадЃЌгУгкХаЖЈвНСЦЦїаЕЙЋЙВЕчЭјЕФБЃЛЄЕФЦРМлРрБ№ЁЃ
ЁЁЁЁИљОнGB 17625.1МАжЦдьЩЬЙцЖЈЕФдЄЦкгУЭОЃЌвНСЦЦїаЕЭЈГЃЗжЮЊAРрЩшБИЁЃШєЙцЖЈЮЊБуаЏЪНЙЄОпРрЩшБИЃЌееУїЩшБИЃЌАќКЌИіШЫМЦЫуЛњЁЂЯдЪОЦїЁЂЕчЪгНгЪеЛњЧвЖдЙЋЙВЙЉЕчЯЕЭГгаЯджјгАЯьЕФЩшБИЃЌПЩЗжБ№ЙцЖЈЮЊBЃЌCЃЌDРрЁЃ
ЁЁЁЁ(вЛ) ЙЄзїФЃЪНбЁШЁддђ
ЁЁЁЁЙЄзїФЃЪНЕФбЁШЁгІЛљгкЖдЗЂЩфЪдбщзюВЛРћддђЃЌФмДњБэВњЦЗе§ГЃЙЄзїЧщПіЯТЕФМЋЖЫзДЬЌЁЃ
ЁЁЁЁ1.ЕЅвЛЙІФм/ФЃЪНвНСЦЦїаЕ
ЁЁЁЁ(1)гІНЋВњЦЗЕФЯћКФЙІТЪЩшжУЕНе§ГЃЪЙгУЕФзюДѓзДЬЌЁЃШчЃКЖдгаФмСПЪфГіЕФЩшБИЃЌЩшжУЪфГіЙІТЪзюДѓЪБВтСПЁЃ
ЁЁЁЁ(2)ШєЩшБИЕФе§ГЃЙЄзїЙ§ГЬЩцМАЖрИіВЛЭЌЕФФЃПщМАЖдгІЙЄзїжмЦкзщГЩЃЌИїФЃПщМАЖдгІЙЄзїжмЦкАДВњЦЗЩшМЦЕФЙЬЖЈЫГађЭъГЩЖЏзїЃЌдђВтСПЪБгІОЁСПКИЧетаЉФЃПщМАЙЄзїжмЦкЁЃШчЃКIVDЩшБИЕФМьВтЗжЮіЙЄзїФЃЪНЩцМАНјбљЁЂЗѕг§ЁЂМьВтЁЂЧхЯДЕШЖрИіФЃПщЃЌИїФЃПщЖдгІЙЬЖЈЙЄзїжмЦкЃЌВтСПЪБгІАќКЌЭъећИіЙЄзїСїГЬЁЃ
ЁЁЁЁ2.ЖрЙІФм/ФЃЪНЕФвНСЦЦїаЕ
ЁЁЁЁ(1)ШєИїЙІФм/ФЃЪНЖРСЂдЫааЃЌРэТлЩЯУПжжЙІФм/ФЃЪНОљгІНјааВтСПЃЌЛђгЩжЦдьЩЬЬсЙЉзюВЛРћЕФвЛжжЛђМИжжЙІФм/ФЃЪНЃЌгІЬсЙЉЯъЯИЕФРэОнжЄЪЕЮЊзюВЛРћЕФЙІФм/ФЃЪНЃЌРэОнАќРЈжЦдьЩЬЬсЙЉЕФММЪѕРэТлЗжЮізЪСЯКЭ/ЛђЪдбщЪ§ОнЁЃ
ЁЁЁЁ(2)ШєИїФЃЪНПЩзщКЯдЫааЃЌбЁдёзюВЛРћЕФзщКЯЗНЪННјааВтСПЃЌгЩжЦдьЩЬЬсЙЉЯъЯИЕФРэОнжЄЪЕЮЊзюВЛРћЕФзщКЯЗНЪНЃЌРэОнАќРЈжЦдьЩЬЬсЙЉЕФММЪѕРэТлЗжЮізЪСЯКЭ/ЛђЪдбщЪ§ОнЁЃ
ЁЁЁЁ(3)ШєФмШЗЖЈЫљгаЙЄзїФЃЪНЩцМАЕФгВМўЕчТЗОљЯрЭЌЃЌдђбЁШЁЙІТЪзюДѓЕФЙЄзїФЃЪНЁЃ
ЁЁЁЁ3.ПЩСЌНгНЛСїЕчЭјЧвДјгаФкВПЕчдДЕФЩшБИ
ЁЁЁЁ(1)ШчСЌНгНЛСїЕчЭјЪБНізїГфЕчзДЬЌЃЌдђжЛашВтСПЦфГфЕчФЃЪНЁЃФкВПЕчдДЙЄзїФЃЪНгІЕЅЖРВтСПЁЃ
ЁЁЁЁ(2)СЌНгНЛСїЕчЭјЪБПЩе§ГЃЙЄзїЃЌдђНЛСїФЃЪНКЭФкВПЕчдДЙЄзїФЃЪНОљашвЊВтСПЁЃСЌНгНЛСїЕчЭјЪБНізїГфЕчзДЬЌЃЌЛЙгІПМТЧГфЕчФЃЪНЁЃ
ЁЁЁЁ4.ЬиЪтРрЩшБИ
ЁЁЁЁ(1)ГЌЩљжЮСЦРрЩшБИашЗжБ№ВтСПШЋЙІТЪЪфГіКЭАыЙІТЪЪфГіФЃЪНЃЌЙЬЖЈЪфГіГ§ЭтЁЃ
ЁЁЁЁ(2)ИпЦЕЪжЪѕРрЩшБИгІСЌНгКУЫљгаИНМўКѓЃЌдкД§ЛњФЃЪНЯТВтСПЁЃ
ЁЁЁЁ(3)Ждгке§ГЃЙЄзїЪБашЭЈЕчЃЌЧвЕЅЖРгЩГЇМвЩњВњЕФгадДвНСЦЦїаЕХфМўВњЦЗ(ШчЃКИпЦЕЪжЪѕЩшБИЕФЕчМЋАхМАЕЖЭЗЁЂЪжЪѕЖЏСІзАжУЕФЪжЛњ)ЃЌвђДјгаЕчзгдЊМўЃЌЪдбщЪБгІСЌНгЕНдЄЦкЪЪХфЕФжїЛњЩЯНјааВтСПЁЃЕЅЖРгЩГЇМвЩњВњЕФЮодДРрХфМўВњЦЗЮоашВтСП(ШчЃКЪжЪѕЖЏСІзАжУЕФЕЖОп)ЁЃ
ЁЁЁЁ(4)Жде§ГЃЪЙгУЪБашХфКЯЦфЫћЩшБИЪЙгУЕФвНСЦЦїаЕЃЌгІЖдвЛИіЛђЖрИіДњБэЭЈГЃЪЙгУЕФЕфаЭХфжУНјааВтСПЃЌШєПЩЭЌЪБСЌНгЪЙгУЃЌдђгІСЌНгЫљгаИЈжњЩшБИКѓНјааВтСПЁЃашСЌНгЪЙгУЕФЮодДРрХфМўЃЌШєСЌНгКѓВЛгАЯьВњЦЗИКдиЃЌдђЮоашСЌНгЃЌШєгАЯьВњЦЗИКдиЃЌдђашСЌНгВтСПЁЃ
ЁЁЁЁ(5)ЖдгкЙЄзїФЃЪНЪфГіжмЦкЯрЖдгкЪдбщЪБМфНЯЖЬЕФЩшБИЃЌгІБЃжЄЪдбщЦЕЖЮИВИЧЭъШЋЃЌПЩЕЅЖРЩшжУЙЄГЬФЃЪНБЃжЄЙЄзїФЃЪНГжајдЫааЁЃ
ЁЁЁЁ(Жў)ЗЂЩфВПЗжЕФЪдбщГЃМћЛэУтЧщПі
ЁЁЁЁ1.ЮоЯпЕчвЕЮёЕФБЃЛЄЪдбщЛэУтЧщПі
ЁЁЁЁ(1)ЙцЖЈНігУгкЦСБЮГЁЫљЕФЩшБИЛђЯЕЭГ
ЁЁЁЁвНСЦЦїаЕзЂВсДњРэЖдгкЙцЖЈНігУгкЦСБЮГЁЫљЕФЩшБИЛђЯЕЭГЃЌЕБдкЪдбщГЁНјааЪдбщЪБЃЌзюЕЭЩфЦЕЦСБЮаЇФмЁЂзюаЁЩфЦЕТЫВЈЫЅМѕЕФММЪѕвЊЧѓТњзуYY 0505жа6.8.3.201c)2)жаЫљЙцЖЈЕФвЊЧѓЃЌGB 4824ЕФЕчДХЗјЩфЩЇШХЯожЕЁЂЕчдДЖЫЩЇШХЕчбЙЯожЕПЩИљОнБъзМЙцЖЈдіМгЁЃ
ЁЁЁЁ(2)КЌгаЮоЯпЕчЩшБИЕФЩшБИКЭЯЕЭГ
ЁЁЁЁЖдгкКЌгавбНјааСЫЪдбщЕФЮоЯпЕчЩшБИЁЂВЂШЯЮЊИУЮоЯпЕчЩшБИЗћКЯЪЪгУЕФЙњМвЮоЯпЕчЗЈЙцЕФЩшБИКЭЯЕЭГЃЌШчЙћЪЪгУЕФЮоЯпЕчЗЈЙцЕФЗЂЩфЯожЕаЁгкЛђЕШгкCISPRЯрЖдгІЕФЙњМвБъзМЕФЕчДХЩЇШХЯожЕЃЌИУЩшБИЛђЯЕЭГПЩУтгкCISPRЯрЖдгІЕФЙњМвБъзМЕФЕчДХЩЇШХвЊЧѓЕФЪдбщЁЃКЌгаЩфЦЕЗЂЩфЛњЕФЩшБИЛђЯЕЭГЃЌдкЗЂЩфЛњЕФзЈгУЗЂЩфЦЕЖЮРяУтгшYY 0505БъзМЕФЗЂЩфвЊЧѓЁЃ
ЁЁЁЁ(3)ДѓаЭгРОУАВзАЕФЩшБИКЭЯЕЭГдкНјааЯжГЁВтСПЪБЃЌПЩУтгшВтСПДЋЕМЗЂЩфЃЌЕЋЛљгкЯжЪЕЪЙгУГЁОАЯТЕФЗчЯеПМТЧЃЌНЈвщВЮееЪдбщГЁЫљЯТЕФЗНЗЈВтСПЁЃ
ЁЁЁЁ(4)ВЛгЩЭјЕчдДЙЉЕчЕФЩшБИКЭЯЕЭГ(ШчЃКФкВПЕчдДЩшБИ)ПЩУтгшЕчдДЖЫЩЇШХЕФВтСПЁЃ
ЁЁЁЁ2.ЙЋЙВЕчЭјЕФБЃЛЄЪдбщЛэУтЧщПі
ЁЁЁЁ(1)дЄЦкВЛСЌНгЙЋЙВЕчЭјЕФЩшБИКЭЯЕЭГПЩУтгкаГВЈЪЇецЁЂЕчбЙВЈЖЏКЭЩСЫИЕФВтСПЁЃ
ЁЁЁЁ(2)УПЯрЖюЖЈЪфШыДѓгк16AЕФвНгУЕчЦјЩшБИКЭЯЕЭГПЩУтгшаГВЈЪЇецЁЂЕчбЙВЈЖЏКЭЩСЫИЕФВтСПЁЃвНгУЕчЦјЩшБИКЭЯЕЭГвдЭтЕФУПЯрЖюЖЈЪфШыДѓгк16AЧвАДGB 4824ЗжЮЊBРрЕФЩшБИ(ШчЃКЪЪгУGB/T 18268ЕФЩшБИ)АДIEC 61000-3-12КЭIEC 61000-3-11ВтСПЁЃ
ЁЁЁЁ(3)ЖюЖЈЙІТЪ75WМАвдЯТЕФЩшБИ(ееУїЩшБИГ§Эт)ЃЌПЩУтгшаГВЈЪЇецЕФВтСПЁЃ
ЁЁЁЁЫФЁЂПЙШХЖШВПЗжЕФММЪѕПМСП
ЁЁЁЁвНСЦЦїаЕВњЦЗзЂВсДњРэвНСЦЦїаЕЕчДХМцШнадЕФПЙШХЖШВПЗжжївЊЛљгкЖдЛљБОадФмКЭЗћКЯадзМдђЕФММЪѕПМСПЁЃ
ЁЁЁЁЛљБОадФмЕФИХФюРДдДгкIEC 60601ЃЌЪЧжИгыЛљБОАВШЋВЛЯрЙиЕФСйДВЙІФмЕФадФмЃЌЦфЩЅЪЇЛђНЕЕЭЕНГЌЙ§жЦдьЩЬЙцЖЈЕФЯожЕЛсЕМжТВЛПЩНгЪмЕФЗчЯеЁЃЮЊЪЕЯждЄЦкЪЙгУЃЌвНгУЕчЦјЩшБИЛђЯЕЭГашЪмЬиЖЈЯожЦЃЌетаЉЯожЦЭЈГЃгЩжЦдьЩЬЙцЖЈЃЌдкIEC 60601БъзМзхжагыIEC 60601-1ЕФВЂСаЛђзЈгУБъзМвВгаЬиЪтЙцЖЈЁЃЮЊСЫЗРжЙЛМепЁЂВйзїепКЭЦфЫћШЫдБЪмЕНЩЫКІЃЌЫљгаЕФЬиеїКЭЙІФмашвЊе§ШЗЬхЯжКЭдЫааЪЧживЊЕФЃЌЕЋВЂЗЧвНгУЕчЦјЩшБИЕФУПвЛЬиеїЛђЙІФмЖМЪЧЛљБОадФмЃЌР§ШчЃКФГаЉжЮСЦРрЩшБИдкжЮСЦЪБЕФвєРжВЅЗХЙІФмЃЌНізїЮЊгщРжгУЭОЁЃЕБдЫааЕФЪЇаЇЛсЕМжТЛМепЁЂВйзїепКЭЦфЫћШЫдБДІгкВЛПЩНгЪмЕФЗчЯеЪБЃЌФЧаЉЬиеїЛђЙІФмБЛПДзїЪЧЛљБОадФмЁЃР§ШчЃК
ЁЁЁЁ——ОзЂЩфБУЪЙгУЕФвЉЦЗЕФе§ШЗЙмРэЃЌШєВЛОЋШЗ/ДэЮѓЙмРэЃЌЛсИјЛМепДјРДВЛПЩНгЪмЕФЗчЯеЁЃ
ЁЁЁЁ——аФЕчЭМЛњ/МрЛЄвЧДгçֿЗХЕчКѓЛжИДЕФФмСІЃЌШєЛжИДЕФЪЇаЇЃЌдђЛсЕМжТвНЛЄШЫдБВЛе§ШЗЕФЯьгІЃЌИјЛМепДјРДВЛПЩНгЪмЕФЗчЯеЁЃ
ЁЁЁЁ——жижЂМрЛЄЛђЪжЪѕМрЛЄЯЕЭГжаБЈОЏЯЕЭГЕФе§ШЗдЫзїЃЌШєВЛе§ШЗ/ШБЪЇБЈОЏаХКХЃЌдђЛсЕМжТвНЛЄШЫдБВЛе§ШЗЕФЯьгІЃЌИјЛМепДјРДВЛПЩНгЪмЕФЗчЯеЁЃ
ЁЁЁЁ——гУгкеяЖЯЕФвНгУЕчЦјЩшБИеяЖЯаХЯЂЕФНсЙће§ШЗадЃЌШчЙћИјГіВЛе§ШЗЕФаХЯЂЛсЕМжТВЛЪЪвЫЕФжЮСЦЗНЗЈЃЌИјЛМепДјРДВЛПЩНгЪмЕФЗчЯеЁЃ
ЁЁЁЁгыЛљБОАВШЋЯрЙиЕФЃЌШчЃКЩшБИЕФОјдЕадФмЃЌВЛБЛФЩШыЛљБОадФмЁЃ
ЁЁЁЁЗћКЯадзМдђгыЛљБОадФмУмЧаЯрЙиЃЌМДХаЖЈЩшБИЛђЯЕЭГЪЧЗёФмЬсЙЉЛљБОадФмВЂБЃГжАВШЋЕФЦРХазМдђЁЃИљОнYY 0505жа36.202.1j)ЕФЙцЖЈЃЌЗћКЯадзМдђАќКЌВЛдЪаэГіЯжЃК
ЁЁЁЁ——ЦїМўЙЪеЯ;
ЁЁЁЁ——ПЩБрГЬВЮЪ§ЕФИФБф;
ЁЁЁЁ——ЙЄГЇФЌШЯжЕЕФИДЮЛ(жЦдьЩЬЕФдЄжУжЕ);
ЁЁЁЁ——дЫааФЃЪНЕФИФБф;
ЁЁЁЁ——ащМйБЈОЏ;
ЁЁЁЁ——ШЮКЮдЄЦкдЫааЕФжежЙЛђжаЖЯЃЌМДЪЙАщгаБЈОЏ;
ЁЁЁЁ——ШЮКЮЗЧдЄЦкдЫааЕФВњЩњЃЌАќРЈЗЧдЄЦкЛђЗЧЪмПиЕФЖЏзїЃЌМДЪЙАщгаБЈОЏ;
ЁЁЁЁ——ЯдЪОЪ§жЕЕФЮѓВюДѓЕНзувдгАЯьеяЖЯЛђжЮСЦ;
ЁЁЁЁ——ВЈаЮЩЯЕФдыЩљЃЌФбвдДгЩњРэВњЩњЕФаХКХжаЧјЗжЃЌЛђепетаЉдыЩљЛсгАЯьЕНЖдЩњРэВњЩњЕФаХКХЕФХаЖЯ;
ЁЁЁЁ——ЭМЯёЩЯЕФЮБгАЛђЪЇецЃЌДЫЮБгАФбвдДгЩњРэВњЩњЕФаХКХжаЧјЗжЛђЪЇецЛсгАЯьЕНЖдЩњРэВњЩњЕФаХКХЕФХаЖЯ
ЁЁЁЁ——здЖЏеяЖЯЛђжЮСЦЩшБИКЭЯЕЭГдкНјааеяЖЯЛђжЮСЦЪБЪЇаЇЃЌМДЪЙАщЫцзХБЈОЏЕШЧщПіЁЃ
ЁЁЁЁЕЋЩшБИКЭЯЕЭГПЩвдГіЯжВЛгАЯьЛљБОадФмКЭАВШЋЕФадФмНЕЕЭЃЌР§ШчЃК
ЁЁЁЁ——гАЯёЯЕЭГЯдЪОЕФПЩФмЪЧБфИќЙ§ЕФЭМЯёЃЌЕЋдкФГжжГЬЖШЩЯВЛЛсЖдеяЖЯЛђепжЮСЦВњЩњгАЯьЁЃ
ЁЁЁЁ——аФТЪМрЛЄвЧЯдЪОЕФаФТЪПЩФмгаДэЮѓЃЌЕЋетИіСПдкСйДВЩЯЮоУїЯдгАЯьЁЃ
ЁЁЁЁ——ЛМепМрЛЄвЧдкВЈаЮЩЯЯдЪОГіЩйСПдыЩљЛђЫВБфЃЌИУдыЩљЛђЫВБфВЛЛсгАЯьеяЖЯЁЂжЮСЦКЭМрЛЄЁЃ
ЁЁЁЁвНСЦЦїаЕзЂВсзЩбЏЬсабФњЪЪгУGB/T 18268БъзМЕФЩшБИЃЌЫфШЛУЛгаЛљБОадФмЖЈвхЃЌЕЋгывНгУЕчЦјЩшБИРрЫЦЃЌПЙШХЖШгІПМКЫгыЩшБИдЄЦкгУЭОЯрЙиЕФЙІФмКЭадФмЁЃШчЃКЬхЭтеяЖЯ(IVD)ЩшБИЃЌВЛгУгкЛМепЮЌГжЩњУќЛђИДЫеЃЌЙЪеЯВЛЛсжБНгЕМжТЛМепЫРЭіЛђбЯжиЪмЩЫЁЃЕЋIVDвНСЦЩшБИжаЕФЙЪеЯПЩФмдьГЩДэЮѓЕФжИЪОжЕЃЌгЩДЫЕМжТДэЮѓЕФжЮСЦеяЖЯЃЌМДЮѓеяЗчЯеЁЃЖдгкФГаЉЗжЮіЮявдМАдкФГаЉЧщПіЯТЃЌДэЮѓЕФНсТлИјЛМепДјРДбЯжиЕФЩЫКІЁЃЖдгкДѓаЭIVDвНСЦЩшБИЃЌЕчДХЩЇШХвВЛсв§Ц№ЙЪеЯЃЌжБНгЭўаВЕНВйзїепЃЌР§ШчЗЧдЄЦкЕФЛњаЕвЦЖЏЁЃЖдЪЪгУGB/T 18268БъзМЕФЩшБИЃЌПЙШХЖШЕФПМКЫгІЙцЖЈЦРЖЈПЙШХЖШЪдбщНсЙћЕФЙІФмКЭ/ЛђадФмЃЌЩЅЪЇКЭНЕЕЭВЛгІЕМжТЪЃгрЗчЯеГЌЙ§ПЩНгЪмЗЖЮЇЁЃдкПЙШХЖШЪдбщЪБЃЌЩшБИГ§ВЛФмЗЂЩњЫ№ЛЕЭтЃЌВЩгУвдЯТЭЈгУадФмХаОнддђЃК
ЁЁЁЁ——адФмХаОнAЃКЪдбщЪБЃЌдкЙцЗЖЯожЕФкадФме§ГЃЁЃ
ЁЁЁЁ——адБ№ХаОнBЃКЪдбщЪБЃЌЙІФмЛђадФмднЪБНЕЕЭЛђЩЅЪЇЃЌЕЋФмздааЛжИДЁЃ
ЁЁЁЁ——адФмХаОнCЃКЪдбщЪБЃЌЙІФмЛђадФмднЪБЩЅЪЇЃЌЕЋашвЊВйзїепИЩдЄЛђЯЕЭГИДЮЛЁЃ
ЁЁЁЁвдIVDЩшБИЮЊР§ЃЌадФмХаОнгІИљОнЪдбщЯюФПШЗЖЈЃЌВЂзлКЯПМТЧПЩФмгАЯьЪ§ОнНсЙћЕФЪмЪдЩшБИЙЄзїФЃЪНКЭПЩФмгАЯьбљЦЗДІРэКЭгУЛЇНгПкЕФЪмЪдЩшБИЙЄзїФЃЪНЁЃЪдбщНсЙћПЩФмБэЯжЮЊадФмХаОнAЁЂBЛђCЃЌЕЋВЛгІЫ№КІЪЙЪЃгрЗчЯеБЃГждкПЩНгЪмЗЖЮЇФкЫљБиаыЕФадФмЬиеїЁЃ
ЁЁЁЁ(вЛ) ЛљБОадФмЕФШЗЖЈзМдђ
ЁЁЁЁЛљБОадФмЭЈГЃЪЧгыдЄЦкгУЭОЯрЙиЕФСйДВКЫаФЙІФмЃЌЩЅЪЇКЭНЕЕЭжБНггАЯьдЄЦкФПЕФЪЕЯжЃЌВњЩњВЛПЩНгЪмЕФЗчЯеЃЌгІИљОнЗчЯеЗжЮіШЗЖЈЁЃАќРЈВЛЯогквдЯТМИжжЧщПіЃК
ЁЁЁЁ1.ГЃЙцвЊЧѓ
ЁЁЁЁФмАДдЄЦкЩшЖЈВЮЪ§ЙЄзїЁЂИїАДМќЙІФме§ГЃЃЌВЛВњЩњЗЧдЄЦкЖЏзїЃЌЙЄзїзДЬЌе§ГЃБэЪОЁЃ
ЁЁЁЁ2.гаФмСПЪфГіЕФгадДжЮСЦРрЩшБИКЭЯЕЭГ
ЁЁЁЁЩшБИЪфГіФмСПЁЂЪфГіЪБМф/ЦЕТЪзМШЗЃЌЖЈЮЛзМШЗЃЌВЛВњЩњЗЧдЄЦкЖЏзїЃЌФмСПМрВтзАжУе§ГЃЙЄзїЁЂФмзМШЗБЈОЏ/ЬсЪОЁЃ
ЁЁЁЁ3.ГЩЯёРрЩшБИКЭЯЕЭГ
ЁЁЁЁЩшБИЪфГіФмСПЁЂЪфГіЪБМф/ЦЕТЪзМШЗЃЌЖЈЮЛзМШЗЃЌВЛВњЩњЗЧдЄЦкЖЏзїЃЌФмСПМрВтзАжУе§ГЃЙЄзїЁЂФмзМШЗБЈОЏ/ЬсЪОЃЌЭМЯёадФмЭъКУЃЌе§ГЃЪЖБ№ЃЌВЛгАЯьеяЖЯЁЃ
ЁЁЁЁ4.еяВьЁЂМрЛЄРрЩшБИКЭЯЕЭГ
ЁЁЁЁЩшБИЪфГіФмСПЁЂЪфГіЪБМф/ЦЕТЪзМШЗЃЌЖЈЮЛзМШЗЃЌВЛВњЩњЗЧдЄЦкЖЏзїЃЌФмСПМрВтзАжУе§ГЃЙЄзїЁЂФмзМШЗБЈОЏ/ЬсЪОЃЌИїЩњРэВЮЪ§ВтСПзМШЗЃЌИїеяВь/МрЛЄЙІФме§ГЃБэЪОЁЃ
ЁЁЁЁ5.зЂЪфРрЩшБИКЭЯЕЭГ
ЁЁЁЁЩшБИЕФСїСППижЦЁЂЮТЖШПижЦзМШЗЃЌзЂЪфОЋШЗЖШВЛНЕЕЭЃЌВЛВњЩњЗЧдЄЦкЖЏзїЃЌФмзМШЗБЈОЏ/ЬсЪОЁЃ
ЁЁЁЁ6.ЯћЖОУ№ОњРргадДЦїаЕ
ЁЁЁЁМгвЉОЋШЗЖШВЛНЕЕЭЃЌФмСПМгдиПижЦзМШЗЃЌзїгУЪБМфПижЦзМШЗЃЌФмзМШЗБЈОЏ/ЬсЪОЁЃ
ЁЁЁЁ7.СйДВМьбщРргадДЦїаЕ
ЁЁЁЁМгбљОЋШЗЖШВЛНЕЕЭЃЌМьВтзМШЗЖШЁЂжиИДТЪВЛНЕЕЭЃЌВЛГіЯжМйбєадЁЂМйвѕадМьВтНсЙћЁЃФмзМШЗБЈОЏ/ЬсЪОЁЃПЩДЅМАЕФЛњаЕВПЗжВЛгІГіЯжЗЧдЄЦкЮЛвЦЁЃ
ЁЁЁЁзЂЃКБОЬѕПюЪЪгУGB/T 18268ЕФЩшБИЦРЖЈПЙШХЖШЪдбщНсЙћЕФЙІФмКЭ/ЛђадФмЕФШЗЖЈЁЃ
ЁЁЁЁ8.УЛгаЛљБОадФмЕФЩшБИКЭЯЕЭГ
ЁЁЁЁЖдгкУЛгаЛљБОадФмЕФгадДЦїаЕЃЌПЩВЮееБОВПЗжГЃЙцвЊЧѓПМКЫЦфЫљгаЙІФмЁЃ
ЁЁЁЁ(Жў) ЙЄзїФЃЪНбЁШЁддђ
ЁЁЁЁЙЄзїФЃЪНЕФбЁШЁгІЛљгкЖдПЙШХЖШЪдбщзюВЛРћддђЃЌФмДњБэВњЦЗе§ГЃЙЄзїЧщПіЯТЕФЕфаЭзДЬЌЁЃ
ЁЁЁЁ1.ЕЅвЛЙІФм/ФЃЪНвНСЦЦїаЕ
ЁЁЁЁАДЕЅвЛЙІФм/ФЃЪНЕФГЃМћЙЄзїзДЬЌВтСПЁЃ
ЁЁЁЁ2.ЖрЙІФм/ФЃЪНЕФвНСЦЦїаЕ
ЁЁЁЁ(1)ШєИїЙІФм/ФЃЪНЖРСЂдЫааЃЌРэТлЩЯУПжжЙІФм/ФЃЪНОљгІНјааВтСПЃЌЛђгЩжЦдьЩЬЬсЙЉзюВЛРћЕФвЛжжЛђМИжжЙІФм/ФЃЪНЃЌгІЬсЙЉЯъЯИЕФРэОнжЄЪЕЮЊзюВЛРћЕФЙІФм/ФЃЪНЃЌРэОнАќРЈжЦдьЩЬЬсЙЉЕФММЪѕРэТлЗжЮізЪСЯКЭ/ЛђЪдбщЪ§ОнЁЃ
ЁЁЁЁ(2)ШєИїЙІФм/ФЃЪНПЩзщКЯдЫааЃЌбЁдёзюВЛРћЕФзщКЯЗНЪННјааВтСПЃЌгЩжЦдьЩЬЬсЙЉЯъЯИЕФРэ ОнжЄЪЕЮЊзюВЛРћЕФзщКЯЗНЪНЃЌРэОнАќРЈжЦдьЩЬЬсЙЉЕФММЪѕРэТлЗжЮізЪСЯКЭ/ЛђЪдбщЪ§ОнЁЃ
ЁЁЁЁ(3)ШчФмШЗЖЈЫљгаЙІФм/ФЃЪНЩцМАЕФгВМўЕчТЗОљЯрЭЌЃЌдђИљОндЄЦкЪЙгУЗНЪНвРДЮЛђЙВЭЌВтСПЁЃ
ЁЁЁЁ3.ПЩСЌНгНЛСїЕчЭјЧвДјгаФкВПЕчдДЕФЩшБИ
ЁЁЁЁ(1)ШчСЌНгНЛСїЕчЭјЪБНізїГфЕчзДЬЌЃЌдђжЛашВтСПЦфГфЕчФЃЪНЃЌГфЕчЪБЩшБИгІВЛВњЩњАВШЋЗНУцЮЃЯеЁЃФкВПЕчдДЙЄзїФЃЪНгІЕЅЖРВтСПЁЃ
ЁЁЁЁ(2)СЌНгНЛСїЕчЭјЪБПЩе§ГЃЙЄзїЃЌдђгІАДYY 0505ЕФвЊЧѓдкЕчПьЫйЫВБфТіГхШКЁЂРЫгПЁЂдкЕчдДЙЉЕчЪфШыЯпЩЯЕФЕчбЙднНЕЁЂЖЬЪБжаЖЯКЭЕчбЙБфЛЏПЙШХЖШЪдбщКѓЛЙгІбщжЄЩшБИЛђЯЕЭГНідкЭјЕчдДЙЉЕчЪБМЬајЙЄзїЕФФмСІЁЃЭЌЪБЛЙгІдкСЌНгНЛСїЕчЭјЪБПМТЧЩшБИдке§ГЃЙЄзїКЭГфЕчЪБВЛВњЩњАВШЋЗНУцЮЃЯеЁЃ
ЁЁЁЁ4.ХфКЯЪЙгУЕФЩшБИбЁдё
ЁЁЁЁ(1)ЖдДјгаЕчзгдЊМўЕФХфМўРрВњЦЗгІСЌНгЪЪХфЕФжїЛњКѓНјааВтСПЁЃЮодДРрХфМўИљОнВњЦЗЬиадбЁдёЪЧЗёашНјааВтСПЃЌШєЮодДРрХфМўЛсгАЯьЩшБИдЫааЛђГЩЮЊПЙШХЖШЪдбщЕчЦНЕФДЋЪфТЗОЖЃЌдђбЁдёСЌНгЛђбЁдёзюВЛРћЕФДЋЪфТЗОЖВтСПЁЃШчЃКЪжЪѕЖЏСІЩшБИХфКЯЪЙгУЕФЮодДРрЕЖОпЃЌПЩФмвђЮЊЕЖОпРраЭВЛЭЌЃЌгАЯьОВЕчЗХЕчВтЪдЕФёюКЯТЗОЖЁЃ
ЁЁЁЁ(2)Жде§ГЃЪЙгУЪБашХфКЯЦфЫћЩшБИЪЙгУЕФвНСЦЦїаЕЃЌгІЖдвЛИіЛђЖрИіДњБэЭЈГЃЪЙгУЕФЕфаЭХфжУНјааВтСПЃЌШєПЩЭЌЪБСЌНгЪЙгУЃЌдђгІСЌНгЫљгаИЈжњЩшБИКѓНјааВтСПЁЃИЈжњЩшБИВЛзїЮЊНсЙЙзщГЩЃЌВЛЕЅЖРПМКЫЁЃ
ЁЁЁЁ(Ш§)ПЙШХЖШВПЗжЕФЪдбщГЃМћЛэУтЧщПі
ЁЁЁЁ1.ОВЕчЗХЕч
ЁЁЁЁЪЉМггкЩшБИЛђЯЕЭГЕФЗЧЕМЕчПЩДЅМАВПМўКЭПЩДЅМАВПМўжаВЛПЩДЅМАЕФЕМЕчВПЗжЕФПеЦјЗХЕчЪдбщЃЌШчЙћСЌНгЦїИННќБъга ЃЌдђИУСЌНгЦїПЩУтгкЪдбщЁЃ
ЁЁЁЁ2.ЩфЦЕЕчДХГЁЗјЩф
ЁЁЁЁ(1)ЙцЖЈНігУгкЦСБЮГЁЫљЕФЩшБИКЭЯЕЭГЃЌШчЙћЩфЦЕЦСБЮаЇФмКЭЩфЦЕТЫВЈЫЅМѕТњзуYY 0505жа6.8.3.201c)2)ЕФвЊЧѓЃЌдђИУПЙШХЖШЪдбщЕчЦНгызюЕЭЩфЦЕЦСБЮаЇФмКЭзюаЁЩфЦЕТЫВЈЫЅМѕЕФЪЪгУЕФЙцЖЈжЕГЩБШР§ЁЃ
ЁЁЁЁ(2)ЮЊЦфдЫааФПЕФЖјНгЪеЩфЦЕЕчДХФмЕФЩшБИКЭЯЕЭГЃЌдкеМгУЦЕДјФк(ШчЃКДјРЖбРЭЈаХ)УтгшЛљБОадФмЕФвЊЧѓЃЌЕЋЩшБИЛђЯЕЭГгІБЃГжАВШЋЁЃ
ЁЁЁЁ(3)ЖдПижЦЁЂМрЪгЛђВтСПЩњРэВЮЪ§ЕФЩшБИЃЌдк2 HzЕїжЦЦЕТЪЯТЪдбщЃЌВЛБидк1 kHzЕїжЦЦЕТЪЯТИНМгЪдбщЁЃ
ЁЁЁЁ(4)НсЙЙЩЯВЛПЩЪЕЯжзгЯЕЭГФЃФтдЫааЕФДѓаЭгРОУадАВзАЩшБИКЭЯЕЭГЃЌПЩУтгкЪдбщГЁЕиЪдбщЃЌгІдкАВзАЯжГЁЛђПЊРЋЪдбщГЁЃЌРћгУГіЯждкЕфаЭНЁПЕМрЛЄЛЗОГжаЕФЩфЦЕдД(ШчЃКЮоЯпЕчЛАЁЂЖдНВЛњЕШ)НјаааЭЪНЪдбщЁЃ
ЁЁЁЁ3.ЕчПьЫйЫВБфТіГхШК
ЁЁЁЁ(1)ЛМепёюКЯЕчРТЁЂаХКХЕчРТЁЂЛЅСЊЕчРТГЄЖШаЁгк3 mВЛЪЪгУИУЯюЪдбщЁЃ
ЁЁЁЁ(2)ЖдгкУЛгаНЛСїКЭжБСїЕчдДЪфШыбЁМўЕФЩшБИКЭЯЕЭГЃЌВЛЪЪгУЕчдДЖЫЕФИУЯюЪдбщЁЃ
ЁЁЁЁ4.РЫгП
ЁЁЁЁ(1)дкГѕМЖЕчдДЕчТЗжаУЛгаРЫгПБЃЛЄзАжУЕФЩшБИКЭЯЕЭГЃЌПЩжЛзі±2 kVЯпЖдЕиКЭ±1 kVЯпЖдЯпЕФЪдбщЁЃгаељвщЪБЃЌЛЙгІЗћКЯЫљЙцЖЈЦфЫћПЙШХЖШЪдбщЕчЦНЕФвЊЧѓЁЃ
ЁЁЁЁ(2)ЖдгкУЛгаШЮКЮНгЕиЛЅСЊЕФIIРрЩшБИКЭЯЕЭГУтгкЯпЖдЕиЪдбщЁЃ
ЁЁЁЁ(3)ЖдгкУЛгаНЛСїКЭжБСїЕчдДЪфШыбЁМўЕФЩшБИКЭЯЕЭГЃЌИУЯюЪдбщВЛЪЪгУЁЃ
ЁЁЁЁ(4)Г§90°КЭ270°ЭтЃЌЕБдЪаэдк0°КЭ180°СНИіЯрНЧЩЯЖМЪдбщЪБЃЌвЊЧѓжЛЪдбщЦфжаЕФвЛИіЁЃ
ЁЁЁЁ5.ЩфЦЕГЁИагІЕФДЋЕМЩЇШХ
ЁЁЁЁ(1)ЙцЖЈНігУгкЦСБЮГЁЫљЕФЩшБИКЭЯЕЭГЃЌШчЙћЩфЦЕЦСБЮаЇФмКЭЩфЦЕТЫВЈЫЅМѕТњзуYY 0505жа6.8.3.201c)2)ЕФвЊЧѓЃЌдђИУПЙШХЖШЪдбщЕчЦНгызюЕЭЩфЦЕЦСБЮаЇФмКЭзюаЁЩфЦЕТЫВЈЫЅМѕЕФЪЪгУЕФЙцЖЈжЕГЩБШР§ЁЃ
ЁЁЁЁ(2)ЮЊЦфдЫааФПЕФЖјНгЪеЩфЦЕЕчДХФмЕФЩшБИКЭЯЕЭГЃЌдкеМгУЦЕДјФк(ШчЃКДјРЖбРЭЈаХ)УтгшЛљБОадФмЕФвЊЧѓЃЌЕЋЩшБИЛђЯЕЭГгІБЃГжАВШЋЁЃ
ЁЁЁЁ(3)дкЕчГиГфЕчЦкМфВЛФмЪЙгУЁЂАќРЈЫљгаСЌНгЕчРТЕФзюДѓГЄЖШдкФкЦфзюДѓГпДчаЁгк1 mЧвЮДгыЕиЁЂЭЈаХЯЕЭГЁЂШЮКЮЦфЫћЩшБИЛђЯЕЭГЛђЛМепЯрСЌЕФФкВПЕчдДЙЉЕчЩшБИЃЌУтгшИУЯюЪдбщЁЃашзЂвтЕФЪЧЃЌаЁгк1 m ЕФЛЅСЊЕчРТзїЮЊФкВПСЌНггУПЩУтгшЪдбщЃЌВЛАќКЌЛМепЕчРТЃЌЛМепЕчРТШдашвЊЪдбщЁЃЕчЮЛОљКтЕМЬхашЪдбщЁЃЪЪгУGB/T 18268ЕФЩшБИЃЌЪфШыЪфГіаХКХПижЦЯпВЛГЌЙ§3 mПЩУтгшЪдбщЁЃ
ЁЁЁЁ6.дкЕчдДЙЉЕчЪфШыЯпЩЯЕФЕчбЙднНЕЁЂЖЬЪБжаЖЯКЭЕчбЙБфЛЏ
ЁЁЁЁ(1)ЖюЖЈЪфШыЙІТЪДѓгк1 kVAЃЌЧвЖюЖЈЪфШыЕчСїаЁгкЛђЕШгкУПЯр16 AЕФЗЧЩњУќжЇГжЩшБИКЭЯЕЭГЃЌжЛвЊЩшБИЛђЯЕЭГБЃГжАВШЋЃЌВЛЗЂЩњзщМўЫ№ЛЕВЂЭЈЙ§ВйзїепИЩдЄПЩЛжИДЕНЪдбщЧАзДЬЌЃЌдђдЪаэЦЋРыЕчбЙднНЕПЙШХЖШЪдбщЕчЦНвЊЧѓЁЃ
ЁЁЁЁ(2)ЖюЖЈЪфШыЕчСїГЌЙ§УПЯр16 AЕФЗЧЩњУќжЇГжЩшБИКЭЯЕЭГЃЌУтгкЕчбЙднНЕЪдбщЁЃ
ЁЁЁЁ(3)жЛвЊЩшБИЛђЯЕЭГБЃГжАВШЋЃЌВЛЗЂЩњзщМўЫ№ЛЕВЂЭЈЙ§ВйзїепИЩдЄПЩЛжИДЕНЪдбщЧАзДЬЌЃЌдђдЪаэЦЋРыЕчбЙжаЖЯПЙШХЖШЪдбщЕчЦНвЊЧѓЁЃЩњУќжЇГжЩшБИЛЙгІЬсЙЉЗћКЯЙњМвКЭЙњМЪБъзМЕФБЈОЏЃЌвдБэУїгыЛљБОадФмгаЙиЕФдЄЦкдЫааЕФжежЙЛђжаЖЯЁЃ
ЁЁЁЁ(4)ЖдгкФкВПЕчдДЩшБИЃЌУтгшИУЯюЪдбщЁЃ
ЁЁЁЁ7.ЙЄЦЕДХГЁ
ЁЁЁЁГ§ЗЧЩшБИКЭЯЕЭГдЄЦкНідквЛИіЦЕТЪЙЉЕчЧјФкЪЙгУЃЌЗёдђашвЊдк50 HzКЭ60 HzСНжжЦЕТЪЩЯНјааЪдбщЁЃ
ЁЁЁЁЮхЁЂЕчДХМцШнадУшЪіЮФЕЕ
ЁЁЁЁЕчДХМцШнадУшЪіЮФЕЕЛљгкYY 0505ЁЖвНгУЕчЦјЩшБИ Ек1-2ВПЗжЃКАВШЋЭЈгУвЊЧѓ ВЂСаБъзМЃКЕчДХМцШн вЊЧѓКЭЪдбщЁЗгшвджЦЖЈЃЌгУгквНгУЕчЦјЩшБИЩъБЈзЂВсЫЭМьЪБЬсНЛЁЃЪєгкВтСПЁЂПижЦКЭЪЕбщЪвгУЕФЕчЩшБИВЮееЪЪгУВПЗжЬсНЛИУЮФЕЕЁЃЕчДХМцШнадУшЪіЮФЕЕАќРЈВњЦЗЕчДХМцШнадАВШЋЬиеїЫЕУїЁЂаЭКХИВИЧЫЕУї(ШчЪЪгУ)ЁЂГаХЕЪщЁЃ
ЁЁЁЁ(вЛ) ВњЦЗЕчДХМцШнадАВШЋЬиеїЫЕУї
ЁЁЁЁ1.ВњЦЗЛљБОаХЯЂ
ЁЁЁЁУїШЗВњЦЗЕФУћГЦЁЂаЭКХЙцИёЁЂШэМўАцБО(ЗЂВМАцБОКЭЭъећАцБО)ЁЂзЂВсШЫКЭзЂВсШЫЕижЗЁЂЩњВњЦѓвЕКЭЩњВњЕижЗЁЂВњЦЗЪЪгУЗЖЮЇЁЃЪЪгУЗЖЮЇжївЊУшЪідЄЦкгУЭОЁЂдЄЦкЪЙгУЛЗОГМАЪЙгУЯожЦЁЂЪЪгУШЫШКЁЃ
ЁЁЁЁ2.ЗжзщЗжРраХЯЂ
ЁЁЁЁАДYY 0505ЕФИНТМCЁЂGB 4824ЁЂGB 17625.1КЭGB 17625.2НсКЯБОжИФЯЕкШ§ВПЗжЙигкЗжзщКЭЗжРрЕФвЊЧѓЃЌУїШЗВњЦЗЕФЗжзщКЭЗжРраХЯЂЃЌВЂЫЕУївРОнЁЃ
ЁЁЁЁ3.ЛљБОадФм
ЁЁЁЁ(1)АДIEC 60601БъзММАБъзМзхНсКЯБОжИФЯЕкЫФВПЗжЛљБОадФмЕФШЗЖЈзМдђЃЌУїШЗВњЦЗЕФЛљБОадФмЁЃЪЪгУGB/T 18268ЕФВњЦЗгІУїШЗЦРЖЈПЙШХЖШЪдбщНсЙћЕФЙІФмКЭ/ЛђадФмЁЃ
ЁЁЁЁ(2)УїШЗЛљБОадФмЕФЪдбщЗНЗЈЁЃШєЛљБОадФмЛђЦРЖЈПЙШХЖШЪдбщНсЙћЕФЙІФмКЭ/ЛђадФмВЩгУММЪѕвЊЧѓжаЕФадФмжИБъЃЌддђЩЯгІгыММЪѕвЊЧѓвЛжТЁЃ
ЁЁЁЁ4.ВњЦЗЕФЙЄзїФЃЪН
ЁЁЁЁ(1)НсКЯВњЦЗЫЕУїЪщЃЌИљОнВњЦЗдЄЦкЪЙгУЕФЙІФмзщКЯЁЂЭЈЕРзщКЯЁЂдЫааЗНЪНЃЌУїШЗВњЦЗЕФЫљгадЫааФЃЪНЁЃАДБОжИФЯЕкШ§КЭЕкЫФВПЗжЙЄзїФЃЪНЕФбЁШЁддђвЊЧѓЃЌУїШЗВњЦЗЗЂЩфЪдбщКЭПЙШХЖШЪдбщЫљВЩгУЕФЙЄзїФЃЪНЃЌВЂЫЕУїбЁШЁвРОнЁЃ
ЁЁЁЁ(2)НсКЯВњЦЗЫЕУїЪщЃЌУїШЗВњЦЗЕФЙЄзїЦЕТЪЁЂЩњРэФЃФтЦЕТЪКЭЯьгІЪБМфЁЃ
ЁЁЁЁ(3)НсКЯВњЦЗЫЕУїЪщЃЌУїШЗВњЦЗЕФЙЉЕчЗНЪН(ШчЃКЕЅЯрНЛСїДјНгЕиЁЂШ§ЯрЮхЯпНЛСїЁЂФкВПЕчдДЕчГиЙЉЕчЕШ)ЃЌВЂУїШЗЙЉЕчВЮЪ§(ЕчбЙЁЂЦЕТЪЁЂЪфШыЙІТЪ/ЪфШыЕчСїЁЂЕчГиРраЭЕШ)ЁЃ
ЁЁЁЁ5.ВњЦЗНсЙЙЫЕУї
ЁЁЁЁ(1)УїШЗВњЦЗЕФНсЙЙзщГЩВЂЬсЙЉЭМЪОУшЪіЃЌгУгкУшЪіВњЦЗгВМўзщГЩВПМў/ФЃПщжЎМфЕФЭтВПСЌНгЙиЯЕЃЌШчЪЪгУЃЌЛЙгІАќРЈбЁХфВПМў/ФЃПщЁЃ
ЁЁЁЁ(2)УїШЗВњЦЗаЭЪНЃЌАќРЈБуаЏЪНЩшБИЁЂЬЈЪНЩшБИЁЂТфЕиЪНЩшБИЁЂгРОУадАВзАЩшБИЁЂAаЭзЈгУЩшБИЕШЁЃ
ЁЁЁЁ(3)ИљОнВњЦЗЬиадЃЌУїШЗВњЦЗЩЇШХдДЃЌАќРЈПЊЙиЕчдДЁЂОЇеёЁЂЪБжгЦЕТЪЁЂЕчЛњЕШЃЌжївЊеыЖдЩфЦЕЗЖЮЇ9 kHzЕН3000 GHzЁЃгІЬсЙЉЩЇШХдДУћГЦЁЂЙЄзїЦЕТЪЁЂЖюЖЈВЮЪ§ЁЂАВзАЮЛжУЁЃ
ЁЁЁЁ(4)АДYY 0505жаЭМA.2ЬсЙЉВњЦЗе§ГЃЙЄзїЪБЕФзюДѓГпДчЃЌвдmm×mm×mmБэЪОЁЃ
ЁЁЁЁ(5)ИљОнВњЦЗгВМўзщГЩВПМў/ФЃПщжЎМфЕФСЌНгЙиЯЕЬсЙЉВњЦЗЕФЭтВПСЌНгЕчРТаХЯЂЃЌАќРЈЕчдДЯпЁЂПижЦЯпЁЂI/OЯпЁЂЛЅСЊЯпЁЂЛМепЕчРТЕШЁЃЕчРТаХЯЂАќРЈУћГЦЁЂЕчРТГЄЖШЁЂЦСБЮЧщПіЁЂгУЭОЁЃЙтЯЫВЛзїЮЊЕчРТПМТЧЃЌЕЋашдкСЌНгЗНЪНжаУшЪіЁЃ
ЁЁЁЁ(6)ЬсЙЉВњЦЗЕФЕчДХМцШнЙиМќдЊЦїМўЧхЕЅЃЌАќРЈдЊЦїМўУћГЦЁЂжЦдьЩЬ/ЩЬБъЁЂаЭКХЛђБрКХЁЂММЪѕВЮЪ§ЁЂАВзАЮЛжУЁЂШЯжЄаХЯЂЁЃ
ЁЁЁЁ(7)ШЗЖЈВњЦЗНсЙЙзщГЩвдЭтЫљашвЊХфКЯЪЙгУЕФВПМўЃЌзїЮЊИЈжњЩшБИЬсЙЉаХЯЂЃЌАќРЈЩшБИБрКХ/ађСаКХЁЂУћГЦЁЂжЦдьЩЬЁЂаЭКХ/ЙцИёЁЃЫЕУїШчаФЕчФЃФтЦїЁЂИпЦЕЕчЕЖЁЂВтЪдЙЄзАЕШЁЃ
ЁЁЁЁ6.ЛэУтЪдбщЧщПіЫЕУї
ЁЁЁЁАДБОжИФЯЕкШ§ЁЂЫФВПЗжЪдбщГЃМћЛэУтЧщПіМАЕчДХМцШнадЯрЙиБъзМвЊЧѓЃЌвдСаБэЗНЪНЬсЙЉЛэУтЯюФПКЭЯрЙивРОнЁЃ
ЁЁЁЁ(Жў) аЭКХИВИЧЫЕУї
ЁЁЁЁЖдЭЌвЛвНСЦЦїаЕзЂВсАьРэЕЅдЊКЌЖрИіаЭКХЕФВњЦЗЃЌгІЬсЙЉаЭКХИВИЧЫЕУїЁЃ
ЁЁЁЁ1.ЙЄзїФЃЪНВювьЫЕУї
ЁЁЁЁЬсЙЉИїаЭКХЙЄзїФЃЪНВювьЖдБШБэЃЌИљОнВњЦЗЕчДХМцШнадАВШЋЬиеїЫЕУїжаВњЦЗЙЄзїФЃЪНвЊЧѓЃЌЬсЙЉВњЦЗдЫааФЃЪНЁЂЙЄзїЦЕТЪЁЂЩњРэФЃФтЦЕТЪКЭЯьгІЪБМфЁЂЙЉЕчЗНЪНКЭЙЉЕчВЮЪ§ЕФЯъЯИЖдБШЁЃ
ЁЁЁЁ2.ВњЦЗНсЙЙВювьЫЕУї
ЁЁЁЁЬсЙЉИїаЭКХВњЦЗНсЙЙВювьЖдБШБэЃЌИљОнВњЦЗЕчДХМцШнадАВШЋЬиеїЫЕУїжаВњЦЗНсЙЙЫЕУївЊЧѓЃЌЬсЙЉВњЦЗНсЙЙзщГЩЁЂВњЦЗаЭЪНЁЂВњЦЗЩЇШХдДЁЂВњЦЗГпДчЁЂЭтВПСЌНгЕчРТ/СЌНгЗНЪНЁЂЙиМќдЊЦїМўЁЂХфКЯЪЙгУЕФВПМўЕФЯъЯИЖдБШЁЃШєФкВПЕчТЗНсЙЙДцдкВювьЃЌЛЙгІЬсЙЉФкВПЕчТЗЭМЕФВювьЖдБШЃЌЖдВювьВПЗжгІЭМЪОБъзЂЁЃ
ЁЁЁЁ(Ш§) ГаХЕЪщ
ЁЁЁЁгІЬсЙЉЕчДХМцШнадАВШЋЬиеїЫЕУїЁЂаЭКХИВИЧЫЕУїЕФецЪЕадЩљУїГаХЕЁЃШєЭЌЪБЫЭМьЭЌвЛаЭКХЖрЬЈбљЦЗНјааАВЙцКЭЕчДХМцШнадаЭЪНЪдбщЃЌЛЙгІЬсЙЉЫЭМьбљЦЗвЛжТадЩљУїГаХЕЁЃЬсЙЉЙиМќдЊЦїМўЁЂСЌНгЕчРТЕФММЪѕЙцИёЪщКЭбљЦЗЭтЙлЁЂЭтВПНгПкЁЂЭтВПБъМЧЁЂФкВПЕчТЗЕФЧхЮњееЦЌзїЮЊГаХЕФкШнЕФжЇГХадВФСЯЁЃ
ЁЁЁЁСљЁЂвНСЦЦїаЕзЂВсжЄЩъЧыВњЦЗзЂВсЕЅдЊгыМьВтЕЅдЊ
ЁЁЁЁ(вЛ) зЂВсЕЅдЊЛЎЗжддђ
ЁЁЁЁгадДвНСЦЦїаЕзЂВсЕЅдЊЛЎЗжжївЊвРОнЁЖвНСЦЦїаЕзЂВсЕЅдЊЛЎЗжжИЕМддђЁЗКЭЯрЙиВњЦЗММЪѕЩѓВщжИЕМддђЁЃЕчДХМцШнадзїЮЊгадДвНСЦЦїаЕЕФживЊММЪѕЦРМлЯюФПЃЌОпгаММЪѕЬиеїЩЯЕФЖРЬиадКЭЖрбљадЃЌгыЁЖвНСЦЦїаЕзЂВсЕЅдЊЛЎЗжжИЕМддђЁЗжаЙцЖЈЕФФкШнДцдкЙиСЊадЁЃвђДЫЃЌдкзЂВсЕЅдЊЛЎЗжЪБЃЌЛЙгІНсКЯВњЦЗЕФЕчДХМцШнадАВШЋЬиеїзлКЯЦРМлЁЃ
ЁЁЁЁ1.ЗжзщЗжРрВЛЭЌЕФгадДвНСЦЦїаЕддђЩЯЛЎЗжЮЊВЛЭЌЕФзЂВсЕЅдЊЁЃЗжзщВЛЭЌЩцМАВњЦЗЕФММЪѕдРэЁЂдЄЦкгУЭОЁЂадФмжИБъЕШВњЦЗВЛЭЌ;ЗжРрВЛЭЌЩцМАВњЦЗЕФдЄЦкЪЙгУЛЗОГВЛЭЌЃЌШчЃКAРрЪЪгУдЄЦкВЛгыЙЋЙВЕчЭјСЌНгЕФЩшБИЃЌBРрЪЪгУАќРЈМвгУЛЗОГдкФкдЄЦкгыЙЋЙВЕчЭјСЌНгЕФЩшБИЁЃ
ЁЁЁЁ2.ЛљБОадФмВЛЭЌЕФгадДвНСЦЦїаЕддђЩЯЛЎЗжЮЊВЛЭЌЕФзЂВсЕЅдЊЁЃИљОнЛљБОадФмЖЈвхЃЌВЛЭЌЕФЛљБОадФмЩцМАСйДВЙІФмЕФВЛЭЌЃЌВњЦЗАВШЋадЁЂгааЇадВЛОпБИЕШЭЌадЁЃ
ЁЁЁЁ3.ЙЄзїЦЕТЪВЛЭЌгАЯьВњЦЗЪЪгУЗЖЮЇЪБЃЌддђЩЯЛЎЗжЮЊВЛЭЌзЂВсЕЅдЊЁЃШчЃКжаЦЕжЮСЦЩшБИКЭЕЭЦЕжЮСЦЩшБИЃЌЙЄзїЦЕТЪгызїгУЛњРэЯрЙиЃЌВЛЭЌзїгУЛњРэЖдгІВЛЭЌЪЪгУЗЖЮЇЁЃИпЦЕЪжЪѕЩшБИЃЌВЛЭЌЕФЪфГіЦЕТЪЭЈГЃЖдгІВЛЭЌЕФИпЦЕЪфГіЕЅдЊЃЌЪфГіЦЕТЪВЛЭЌЖдгІВЛЭЌЕФЪжЪѕгУЭОЁЃ
ЁЁЁЁ4.ШєЩшБИЩЇШХдДЕФЦЕТЪВЛгАЯьЪЪгУЗЖЮЇЪБЃЌПЩЛЎЗжЮЊЭЌвЛзЂВсЕЅдЊЁЃШчЃКПижЦаОЦЌЕФЙЄзїЦЕТЪВЛЭЌЃЌЖдЪ§ОнДІРэНіДцдкаЇТЪЩЯЕФЧјБ№ЃЌВЛгАЯьСйДВЙІФмЪЕЯжЃЌПЩЛЎЗжЮЊЭЌвЛзЂВсЕЅдЊЁЃ
ЁЁЁЁ5.ВЛЭЌЙЉЕчЗНЪНЕМжТЪЙгУЗНЪНКЭЪЙгУШЫдБВЛЭЌЪБЃЌддђЩЯЛЎЗжЮЊВЛЭЌзЂВсЕЅдЊЁЃЙЉЕчЗНЪНВЛЭЌЭЈГЃдьГЩВњЦЗЕФНсЙЙзщГЩЁЂаЭЪНВЛЭЌЃЌВЛЭЌЕФВњЦЗНсЙЙКЭаЭЪНЖдгІВЛЭЌЕФЪЙгУЗНЪНКЭЪЙгУШЫдБЁЃШчЃКШ§ЯрЮхЯпНЛСїКЭФкВПяЎЕчГиЙЉЕчЕФжЮСЦЩшБИЃЌВњЦЗНсЙЙЩЯгаНЯДѓВювьЃЌаЭЪНЩЯЗжБ№ЮЊгРОУадАВзАКЭБуаЏЪжГжЪНЃЌдкЪЙгУжаЃЌЖдВйзїШЫдБЁЂЮЌЛЄБЃбјШЫдБЁЂАВзАЪЙгУЛЗОГОљгаВЛЭЌвЊЧѓЃЌЩшБИдкЪЕМЪЪЙгУжаЕФАВШЋЗчЯеВЛОпгаЕШЭЌадЁЃ
ЁЁЁЁ6.ВњЦЗгВМўзщГЩВПМў/ФЃПщжЎМфЕФСЌНгЗНЪНВЛЭЌгАЯьВњЦЗЪЪгУЗЖЮЇЪБЃЌддђЩЯЛЎЗжЮЊВЛЭЌзЂВсЕЅдЊЁЃШчЃКгаЯпСЌНгКЭЮоЯпСЌНгЕФМрЛЄвЧЃЌгаЯпСЌНгЕФМрЛЄвЧЭЈГЃгУгкДВХдМрЛЄЃЌдкЯдЪОЦСЩЯЙлВьЛМепЪ§ОнЃЌЮоЯпСЌНгЕФМрЛЄвЧЭЈГЃПЩдЖГЬМрЛЄЃЌЭЈЙ§ЮоЯпДЋЪфЪ§ОнЕНжааФМрЛЄЦНЬЈдЖГЬЖСШЁЪ§ОнЃЌЪЪгУЗЖЮЇВЛЭЌЃЌВЛгІЛЎЗжЮЊЭЌвЛзЂВсЕЅдЊЁЃ
ЁЁЁЁ7.ХфКЯЪЙгУЕФВПМўВЛЭЌгАЯьВњЦЗЪЪгУЗЖЮЇЪБЃЌддђЩЯЛЎЗжЮЊВЛЭЌзЂВсЕЅдЊЁЃШчЃКЪжЪѕЖЏСІЩшБИХфБИЪЪгУгкећШнЪжЪѕЕФВњЦЗВПМўКЭЪЪгУгкЙЧПЦЪжЪѕЕФВњЦЗВПМўЃЌЕМжТВњЦЗЪЪгУЗЖЮЇВЛЭЌЃЌВЛФмЛЎЮЊЭЌвЛвНСЦЦїаЕВњЦЗзЂВсАьРэЕЅдЊЁЃЕЋЪжЪѕЖЏСІЩшБИХфБИНігУгкЙЧПЦЪжЪѕЕФВЛЭЌзъФЅЕЖОпВПМўЃЌПЩЛЎЗжЮЊЭЌвЛзЂВсЕЅдЊЁЃ
ЁЁЁЁ(Жў) МьВтЕЅдЊЛЎЗжддђ
ЁЁЁЁМьВтЕЅдЊЪЧжИЭЌвЛзЂВсЕЅдЊФкгУгкМьВтЕФДњБэВњЦЗЁЃДњБэВњЦЗЕФбЁШЁЪЧгУгкЖдЭЌвЛзЂВсЕЅдЊФкЖрИіаЭКХВњЦЗЕФМьбщИВИЧЁЃвђДЫЃЌДњБэВњЦЗгІОпБИдкЕчТЗдРэЁЂНсЙЙзщГЩЁЂЙІФм/ФЃЪНЁЂЙЉЕчЗНЪНЁЂЛљБОадФмЕШЗНУцАДзюВЛРћддђЕФМьбщЕфаЭадЁЃ
ЁЁЁЁЭЈГЃбЁШЁБОзЂВсЕЅдЊжаНсЙЙзюИДдгЁЂЙІФм/ФЃЪНзюШЋУцЕФаЭКХЮЊДњБэВњЦЗЃЌашзЂвтНсЙЙЁЂЙІФм/ФЃЪНЕФЩОМѕЖдгкЕчДХМцШнадЕФгАЯьЃЌЛЙгІвдБОжИФЯЕкШ§ЁЂЫФВПЗжЕФЙЄзїФЃЪНбЁШЁддђЮЊвРОнЃЌШЗЖЈЪЧЗёашдіМгЯргІЕФЦфЫћаЭКХвЛЦ№зїЮЊДњБэВњЦЗВЂбЁдёМьбщЯюФПЁЃР§ШчЃК
ЁЁЁЁ——ФГЩшБИАќКЌAКЭBСНИіаЭКХЃЌAаЭКХНсЙЙДјгаНгЕизАжУЃЌBаЭКХМѕЩйСЫНгЕизАжУЃЌВЛФмЫЕУїAаЭКХБШBаЭКХИќОпгаЕфаЭадЁЃ
ЁЁЁЁ——ФГЩшБИАќКЌAКЭBСНИіаЭКХЃЌAаЭКХОпгаa+b+cШ§ИіЙІФмЃЌBаЭКХОпгаa+bСНИіЙІФмЃЌЕЋBаЭКХЕФaЙІФмНЯAаЭКХЕФaЙІФмЪфГіЙІТЪДѓЃЌВЛФмЫЕУїAаЭКХБШBаЭКХИќОпгаЕфаЭадЁЃ
ЁЁЁЁЖдгкШБЩйБивЊЕФММЪѕРэТлЗжЮіКЭ/ЛђЪдбщЪ§ОнзїЮЊРэОнЕФЧщПіЃЌЕчДХМцШнадМьбщгІЕБКИЧЩъБЈЕЅдЊжаЕФШЋВПаЭКХЁЃ
ЁЁЁЁЦпЁЂвНСЦЦїаЕВњЦЗзЂВсжЄЩъБЈзЪСЯвЊЧѓ
ЁЁЁЁБОВПЗжжївЊеыЖдВњЦЗзЂВсЩъБЈЪБЃЌЖдЕчДХМцШнадВПЗжашвЊЬсНЛЕФзЪСЯвЊЧѓЃЌЮДЬсМАЕФзЂВсЩъБЈзЪСЯЦфЫћФкШнгІЗћКЯЁЖЙигкЙЋВМвНСЦЦїаЕзЂВсЩъБЈзЪСЯвЊЧѓКЭХњзМжЄУїЮФМўИёЪНЕФЙЋИцЁЗЕФвЊЧѓЁЃ
ЁЁЁЁ(вЛ) ВњЦЗзЂВс
ЁЁЁЁ1.вНСЦЦїаЕАВШЋгааЇЛљБОвЊЧѓЧхЕЅ
ЁЁЁЁАВШЋгааЇЛљБОвЊЧѓЧхЕЅжаB5.2.3ЁЂB9.5гІЪЪгУЃЌВЂЫЕУїжЄУїЗћКЯадВЩгУЕФЗНЗЈКЭЮЊЗћКЯадЬсЙЉПЭЙлжЄОнЕФЮФМўЁЃ
АВШЋгааЇЛљБОвЊЧѓЧхЕЅЕчДХМцШнадЯрЙиЬѕПюЪОР§
|
ЬѕПюКХ |
вЊЧѓ |
ЪЪгУ |
жЄУїЗћКЯадВЩгУЕФЗНЗЈ |
ЮЊЗћКЯадЬсЙЉПЭЙлжЄОнЕФЮФМў |
|
B5.2.3 |
гыКЯРэПЩдЄМћЕФЭтВПвђЫиЛђЛЗОГЬѕМўгаЙиЕФЗчЯеЃЌБШШчДХГЁЁЂЭтВПЕчДХаЇгІЁЂОВЕчЗХЕчЁЂеяЖЯКЭжЮСЦДјРДЕФЗјЩфЁЂбЙСІЁЂЪЊЖШЁЂЮТЖШвдМАбЙСІКЭМгЫйЖШЕФБфЛЏЁЃ |
ЪЧ
|
ЭЈЙ§зЂВсМьбщКЯИё |
зЂВсМьбщБЈИц
ЃЈБЈИцБрКХЃКXXXXXЃЉ |
|
B9.5 |
вНСЦЦїаЕЕФЩшМЦКЭЩњВњЃЌгІЕБОпгаМѕЩйВњЩњЕчДХИЩШХЕФЗНЗЈЁЃ |
ЪЧ |
ЭЈЙ§зЂВсМьбщКЯИё |
зЂВсМьбщБЈИц
ЃЈБЈИцБрКХЃКXXXXXЃЉ |
ЁЁ2.злЪізЪСЯЁЁЁЁ
(1)ЕчДХМцШнадЯрЙиЕФВњЦЗНсЙЙзщГЩЭМжївЊгУгкУшЪіВњЦЗгВМўзщГЩВПМў/ФЃПщЁЂбЁХфВПМў/ФЃПщжЎМфЕФЭтВПСЌНгЙиЯЕЃЌПЩгыВњЦЗЩъБЈЬсЙЉЕФећЬхНсЙЙЭМВЛЭЌЃЌЭЈГЃВЛвЊЧѓЬхЯжВњЦЗФкВПНсЙЙЁЃбљЦЗСЌНгЭМЪОР§(ЪжЪѕЖЏСІЩшБИ)ЃК
ЁЁЭМ2 ПижЦжїЛњгыШЁЦЄ/РЉЦЄЕЅдЊКЭНХЬЄПЊЙиЭЈЙ§ЕчРТгаЯпСЌНг
ЁЁЁЁ(2)ВњЦЗЕФЪЪгУЗЖЮЇЁЂдЄЦкЪЪгУЛЗОГЁЂЪЪгУШЫШКМАЪЙгУЯожЦгІгыЕчДХМцШнадЕФЗжзщЗжРрЖдгІЕФЕчДХЛЗОГЯрЪЪгІЁЃ
ЁЁЁЁ(3)ВњЦЗдЄЦкгыЦфЫћвНСЦЦїаЕЛђЭЈгУВњЦЗзщКЯЪЙгУЫЕУїжаЃЌгІАќКЌГ§ЕчДХМцШнадВтЪдЫљашИЈжњЩшБИжаВтЪдЩшБИ/ЙЄзАвдЭтЃЌЪЕЯжВњЦЗЙІФмЯрЙиЕФЦфЫћЩшБИ/ВПМўЁЃ
ЁЁЁЁ3.баОПзЪСЯ
ЁЁЁЁдкВњЦЗадФмбаОПжаЃЌЬсЙЉБОжИФЯЕкЮхВПЗжвЊЧѓЕФЕчДХМцШнадУшЪіЮФЕЕЁЃ
ЁЁЁЁ4.ЩњВњжЦдьаХЯЂ
ЁЁЁЁВњЦЗЕчДХМцШнадВтЪджаЃЌЭЈГЃЩцМАВњЦЗКЯЙцадећИФЃЌВњЦЗФкВПНсЙЙКЭЩњВњЙЄвеЙ§ГЬПЩФмДцдквЛЖЈГЬЖШЕїећЁЃШчЪЪгУЃЌгІЬсЙЉВњЦЗећИФКѓЕФЩњВњЙЄвеСїГЬЭМКЭЙ§ГЬПижЦЕуЃЌВЂЬсЙЉећИФЧАКѓЕФЙЄвеЖдБШЫЕУїЁЃ
ЁЁЁЁ5.ВњЦЗЗчЯеЗжЮізЪСЯ
ЁЁЁЁЗчЯеЗжЮіжагІАќКЌЖдЕчДХФмСПЮЃЯе(дД)ЕФЗжЮіЃЌАДееYY/T 0316жаИНТМEЕФЪОР§ЃЌУшЪіЮЃЯе(дД)ЁЂПЩдЄМћЕФЪТМўађСаЁЂЮЃЯеЧщПіКЭПЩЗЂЩњЕФЩЫКІжЎМфЕФЙиЯЕЁЃЪОР§ШчЯТЃК
ВњЦЗЮЃЯеЃЈдДЃЉЁЂПЩдЄМћЕФЪТМўађСаЁЂ
ЮЃЯеЧщПіКЭПЩЗЂЩњЕФЩЫКІжЎМфЕФЙиЯЕЪОР§
|
ЮЃЯеЃЈдДЃЉ |
ПЩдЄМћЕФЪТМўађСа |
ЮЃЯеЧщПі |
ЩЫКІ |
ЕчДХФмСП
ЃЈЕчДХИЩШХЃЉ |
ЪжЪѕЪвФкЦфЫћЩшБИЖдЙЧзщжЏЪжЪѕЩшБИЕчДХИЩШХЕМжТЕчПиВПМўЗЧПижЦЦєЖЏЁЂдЫзЊЃЛ
ЙЧзщжЏЪжЪѕЩшБИИЩШХЦфЫћЪжЪѕЩшБИЕФе§ГЃЙЄзїЁЃ |
ЩшБИЛюЖЏВПМўвтЭтдЫЖЏЃЛЩшжУВЮЪ§здааИФБфЃЛ
ЦфЫћЭЌЪБЪЙгУЕФМрЛЄЛђЩњУќЮЌГжЯЕЭГЮоЗЈе§ГЃЙЄзїЁЃ |
ЛМепЛњаЕЫ№ЩЫЁЂЫРЭіЃЛ
МфНгЕМжТЛМепЫРЭіЁЃ |
ЛЏбЇЗЂЙтУтвпЗжЮіЩшБИЖдЪмЕНЪЙгУЛЗОГжаЦфЫћЩшБИЕчДХИЩШХЕМжТбљЦЗДІРэФЃПщВЛЙЄзїЁЃ
ЛЏбЇЗЂЙтУтвпЗжЮіЩшБИИЩШХЪЙгУЛЗОГжаЕФЦфЫћIVDЩшБИ |
ВЩМЏДэЮѓЕФСйДВМьбщЪ§ОнЁЃ
ЦфЫћIVDЩшБИМгШШФЃПщЮТЖШПижЦзАжУЪЇаЇ |
ЮѓеяЃЌбгЮѓЛМепжЮСЦЃЌМфНгЕМжТЛМепНЁПЕЫ№КІЃЛ
ЕМжТВйзїепЬЬЩЫЁЃ
|
ЁЁЗчЯеЗжЮіжаЛЙгІАќКЌЖдВњЦЗЛљБОадФмЕФЗжЮіЦРЖЈЃЌШЗЖЈгыЛљБОадФмЖдгІЕФЗћКЯадзМдђЁЃЖдGB/T 18268ЕФЩшБИЃЌШчЃКЬхЭтеяЖЯ(IVD)ЩшБИЃЌЛЙгІАДееYY/T 0316ЕФИНТМHЖдДэЮѓЕФМьВщНсТлдьГЩЕФЗчЯеНјааЦРЙРЁЃЗчЯеЗжЮіЪфГігІАќКЌЖдЦРЖЈПЙШХЖШЪдбщНсЙћЕФЙІФмКЭ/ЛђадФмКЭЗћКЯадХаОнЁЃЁЁЁЁ
6.вНСЦЦїаЕзЂВсжЄДњРэВњЦЗММЪѕвЊЧѓ
ЁЁЁЁ(1)ВњЦЗММЪѕвЊЧѓе§ЮФВПЗжгІдкадФмжИБъВПЗжУїШЗЕчДХМцШнадЕФЗжзщЗжРрЁЂВњЦЗЪЪгУЕФЕчДХМцШнадМьбщБъзМ(YY0505ЁЂGB/T 18268ЕШ)ЁЂВњЦЗЯрЙижИЕМддђЕФЕчДХМцШнадвЊЧѓЕШЁЃдкМьбщЗНЗЈВПЗжУїШЗЕчДХМцШнадЕФЪдбщЗНЗЈЃЌАќРЈЗЂЩфКЭПЙШХЖШЕФЪдбщЗНЗЈЃЌЦфжаПЙШХЖШЕФЪдбщЗНЗЈгІЫЕУїЖдЛљБОадФмбщжЄЕФЗНЗЈ;ЖдЪЪгУGB/T 18268ЕФЩшБИЃЌгІЫЕУїПЙШХЖШЪдбщЕФадФмХаОнКЭЦРЖЈПЙШХЖШЪдбщНсЙћЕФЙІФмКЭ/ЛђадФмЕФЪдбщЗНЗЈЁЃ
ЁЁЁЁ(2)ВњЦЗММЪѕвЊЧѓИНТМВПЗжгІАќКЌБОжИФЯЕкЮхВПЗжжаВњЦЗЕчДХМцШнадАВШЋЬиеїЫЕУїжагыМьбщЕФЯрЙиФкШнЁЃИНТМжаНЈвщСаБэЬхЯжЕчДХМцШнадБъзМжаМьбщЬѕПюЪЪгУЧщПіЁЃ
ЁЁЁЁ(3)ВњЦЗММЪѕвЊЧѓдЄЦРМлвтМћжагІЬхЯжЕчДХМцШнадМьбщБЈИцБрКХЁЂВЩБъЧщПі(СаГіЪЪгУБъзМКХ)ЁЂДњБэадВњЦЗбЁШЁМАаЭКХИВИЧЫЕУїЕФаХЯЂ(ШчЪЪгУ)ЁЃМьбщЛњЙЙЖдЦѓвЕЬсЙЉЕФЕчДХМцШнадУшЪіЮФЕЕНсКЯВњЦЗЪЕМЪЧщПізлКЯЦРЙРШЗШЯКѓЃЌПЩжБНгГіОпДњБэадВњЦЗбЁШЁМАаЭКХИВИЧЕФНсТлвтМћЃЌИУвтМћврПЩдкМьбщБЈИцЪзвГБИзЂРИжаУшЪіЁЃ
ЁЁЁЁ7.ВњЦЗзЂВсМьбщБЈИц
ЁЁЁЁЕчДХМцШнадВњЦЗзЂВсМьбщБЈИцгІАДееЁЖЙњМвЪГЦЗвЉЦЗМрЖНЙмРэОжАьЙЋЬќЙигкгЁЗЂYY 0505МьбщБЈИцВЮПМИёЪНЕФЭЈжЊЁЗ(ЪГвЉМрАьаЕЙм[2013]29КХ)ЕФвЊЧѓГіОпЁЃЛЙгІАќКЌвдЯТФкШнЃК
ЁЁЁЁ(1)МьбщБЈИцееЦЌвГЕФбљЦЗЭтВПЭМЦЌгІЬхЯжНсЙЙзщГЩ(КЌбЁХфМў)ЁЂСЌНгЗНЪН/СЌНгЕчРТЁЂЭтВПНгПкЁЂЭтВПБъМЧЁЂУњХЦЁЂКЌШэМўАцБОаХЯЂЕФНчУцЕШЭтВПАВШЋЬиеїЪЕЮяЭМЁЃЭтВПЭМЦЌЛЙгІАќКЌИЈжњЩшБИаХЯЂЁЃ
ЁЁЁЁ(2)бљЦЗФкВПНсЙЙЭМЦЌгІЬхЯжбљЦЗЕФФкВПЕчТЗВМОжЁЂСЌНгЁЂгАЯьЕчДХМцШнадЕФживЊЙиМќдЊЦїМўЁЂжївЊЩЇШХдДЕШФкВПАВШЋЬиеїЪЕЮяЭМЁЃ
ЁЁЁЁ(3)ШчЪЪгУЃЌбљЦЗЭМЦЌгІЬхЯжЕчДХМцШнадМьВтжаЕФећИФЧщПіЃЌАќКЌдіМг/ИќЛЛЕФдЊЦїМўМААВзАЮЛжУЁЂдіМгЕФЦСБЮКЭ/ЛђНгЕиДыЪЉЁЂИќИФЕФФкВПзпЯп/СЌНгЗНЪНЕШЭМЦЌУшЪіЃЌБивЊЪБЃЌПЩдіМгЮФзжУшЪіЁЃ
ЁЁЁЁ(4)ЖдАВЙцКЭЕчДХМцШнадМьбщжаЗЂЩњећИФЕФЧщПіЃЌМьбщЛњЙЙгІЙизЂећИФЯюЃЌЦРЙРКѓЪЕЪЉБивЊЕФЙиСЊадВЙГфМьбщВЂГіОпВЙГфМьбщБЈИцЁЃВЙГфМьбщБЈИцгІдкдЄЦРМлвтМћЛђМьбщБЈИцБИзЂРИжаЬхЯжАВЙцКЭЕчДХМцШнадМьбщЕФЙиСЊадЁЃ
ЁЁЁЁ(5)вРОнЁЖЙигкYY 0505-2012вНСЦЦїаЕаавЕБъзМЪЕЪЉгаЙиЙЄзївЊЧѓЕФЭЈжЊЁЗ(ЪГвЉМрАьаЕ[2012]151КХ)ЮФМўвЊЧѓЃЌЖдЕчДХМцШнБъзМЪЕЪЉЗжАќМьВтЕФвНСЦЦїаЕМьВтЛњЙЙЃЌдкЪеЕНЗжАќЗНЩЯЪіИёЪНМьбщБЈИцКѓЃЌгІАДЩЯЪівЊЧѓЩѓКЫШЗШЯ(БивЊЪБЃЌгІНјааЙиСЊадВЙГфМьбщ)ЃЌВЂГіОпАќКЌЕчДХМцШнадФмЕФЭъећаЭЪНМьбщБЈИцЁЃ
ЁЁЁЁ(6)ЪЪгУGB/T 18268ЕФВњЦЗЃЌМьбщБЈИцжаЛЙгІЬхЯжИїМьбщЯюФПЕФадФмХаОнЁЃ
ЁЁЁЁ8.ВњЦЗЫЕУїЪщ
ЁЁЁЁВњЦЗЫЕУїЪщжагІАќКЌЕчДХМцШнБъзМжаЖдЫЕУїЪщЕФвЊЧѓЃЌНЈвщЩшжУЖРСЂеТНкНјааБраДЁЃвНгУЕчЦјЩшБИКЭЯЕЭГгІАДYY 0505ЕФ6.8ЫцЛњЮФМўБраДЕчДХМцШнадЕФЫЕУїЪщФкШнЃЌЪЪгУGB/T 18268ЕФвНСЦЦїаЕгІАДGB/T 18268ЯЕСаБъзМЕФЕк9еТЪЙгУЫЕУїБраДЕчДХМцШнадЕФЫЕУїЪщФкШнЁЃ
ЁЁЁЁ(1)вНгУЕчЦјЩшБИКЭЯЕЭГ
ЁЁЁЁYY 0505ЕФ6.8ЫцЛњЮФМўАќКЌ6.8.2.201ЪЙгУЫЕУїЪщКЭ6.8.3.201ММЪѕЫЕУїЪщСНВПЗжЁЃ
ЁЁЁЁЪЙгУЫЕУїЪщжаЃЌашзЂвт ЗћКХЕФЪЪгУвЊЧѓКЭЪЙгУЯожЦЃЌгІдкЫЕУїЪщжаУїШЗЬхЯжЁЃ
ЁЁЁЁММЪѕЫЕУїЪщжаЃЌашзЂвтYY 0505жаБэ201жСБэ208жИФЯКЭжЦдьЩЬЕФЩљУїЕФЪЪгУадЃЌгІБъзМвЊЧѓбЁдёЪЪгУЕФБэИёЬюаДЃЌдкЫЕУїЪщжаУїШЗЬхЯжЁЃБэ201ЖдгІЕчДХЗЂЩфВПЗжЃЌЪЪгУЫљгаЩшБИКЭЯЕЭГЃЌПЩАДБъзМжаЭМ201КЭЭМ202ЕФжИв§НјааЬюаД;Бэ202жСБэ208ЖдгІЕчДХПЙШХЖШВПЗжЃЌЦфжаБэ202ЪЪгУЫљгаЩшБИКЭЯЕЭГЃЌПЩАДБъзМжаЭМ203ЕФжИв§НјааЬюаД;Бэ203ЁЂ205ЪЪгУЩњУќжЇГжЩшБИКЭЯЕЭГЃЌБэ204ЁЂ206ЪЪгУЗЧЩњУќжЇГжЩшБИКЭЯЕЭГЃЌПЩЗжБ№АДЭМ204КЭЭМ205ЕФжИв§НјааЬюаД;Бэ207ЁЂ208ЗжБ№ЪЪгУЙцЖЈНігУгкЦСБЮГЁЫљЕФЩњУќжЇГжЩшБИКЭЯЕЭГгыЗЧЩњУќжЇГжЩшБИКЭЯЕЭГЁЃ
ЁЁЁЁ(2)ЪЪгУGB/T 18268ЕФвНСЦЦїаЕ
ЁЁЁЁЪЪгУGB/T 18268ЕФвНСЦЦїаЕЕФЫЕУїЪщвЊЧѓдкзЈгУБъзМЕФЬиЪтвЊЧѓжаЬхЯжЃЌШчGB/T 18268.26ЁЖВтСПЁЂПижЦКЭЪЕбщЪвгУЕФЕчЩшБИ ЕчДХМцШнадвЊЧѓ Ек26ВПЗжЃКЬхЭтеяЖЯ(IVD)вНСЦЩшБИЁЗЃЌЦфжаЕк9еТЪЙгУЫЕУїАќРЈ9.101ЖдIVDЩшБИЪЙгУЫЕУїЕФвЊЧѓЁЂ9.102здМьгУIVDЩшБИЕФЪЙгУЫЕУїЁЂ9.103зЈвЕIVDЩшБИЕФЪЙгУЫЕУїЃЌЗЧздМьгУЁЃЦфЫћЪЪгУGB/T 18268ЕФвНСЦЦїаЕЫЕУїЪщФкШнПЩВЮееGB/T 18268.26ЕФ9.101КЭ9.103БраДЁЃ
ЁЁЁЁ9.ЗћКЯадЩљУї
ЁЁЁЁгІдкЗћКЯБъзМЕФЧхЕЅжаЩљУїЩъБЈВњЦЗашЗћКЯЕФЕчДХМцШнадЭЈгУБъзМЁЂзЈгУБъзМЁЂВњЦЗжИЕМддђЁЃ
ЁЁЁЁ(Жў)аэПЩЪТЯюБфИќ
ЁЁЁЁ1.БфИќЧщПіЩљУї
ЁЁЁЁгІЯъЯИЩљУїВњЦЗММЪѕвЊЧѓЁЂаЭКХЙцИёЁЂНсЙЙзщГЩЁЂдЄЦкЪЙгУЛЗОГЁЂЪЪгУШЫШКЁЂВйзїепЁЂЪЙгУЗНЪНЁЂЪЙгУЯожЦЕФБфЛЏЁЃ
ЁЁЁЁ2.БфИќЩъЧыЯюФПЩъБЈзЪСЯвЊЧѓ
ЁЁЁЁИљОнБфИќЧщПіЩљУїЃЌЬсЙЉЯъЯИБфИќЖдБШБэЃЌБивЊЪБЃЌПЩХфЪОвтЭМКЭЭМЦЌУшЪіЁЃЬсЙЉЕчДХМцШнадУшЪіЮФЕЕЃЌЬхЯжгыВњЦЗЕчДХМцШнадЯрЙиЕФБфЛЏФкШнЃЌгІзЂУїЮДЗЂЩњБфЛЏЕФВПЗжЁЃ
ЁЁЁЁ3.гыВњЦЗБфЛЏЯрЙиЕФАВШЋЗчЯеЙмРэБЈИц
ЁЁЁЁгІИљОнБфЛЏЧщПіжиаТЦРЙРВњЦЗЕФЕчДХФмСПЮЃЯе(дД)ЃЌВЂЬсЙЉБфЛЏКѓЕФЗчЯеЙмРэБЈИцЁЃ
ЁЁЁЁ4.еыЖдВњЦЗММЪѕвЊЧѓБфЛЏВПЗжЕФзЂВсМьбщБЈИц
ЁЁЁЁ(1)діМгВњЦЗаЭКХЃЌдаЭКХЮоБфЛЏ
ЁЁЁЁгІЬсНЛдіМгаЭКХЕФЕчДХМцШнадМьбщБЈИцЁЃПЩНсКЯвНСЦЦїаЕЪзДЮзЂВсЩъБЈЪБЕФЕчДХМцШнадБЈИцЖдаТдіаЭКХЕФВювьадзїЦРЙРЃЌГіОпаТдіаЭКХЕФВЙГфМьбщБЈИцЃЌВЂгыЪзДЮзЂВсЩъБЈЪБЕФЕчДХМцШнадБЈИцећЬхЙиСЊЁЃДцдкЖрИіаТдіаЭКХЃЌгІГіОпЛљгкаТдіаЭКХЕФИВИЧвтМћЃЌМьбщЛњЙЙвВПЩбЁдёЖддаЭКХЕчДХМцШнадзЂВсМьбщБЈИцЦРЙРКѓНјаааТдіаЭКХЕФВювьЛЏВЙГфМьбщЃЌВЂГіОпећЬхИВИЧвтМћЁЃгІдкММЪѕвЊЧѓдЄЦРМлБэЛђВЙГфМьбщБЈИцБИзЂРИжаЬхЯжЁЃ
ЁЁЁЁБфЛЏВПЗжЕФМьбщддђЩЯгІдкдМьбщЛњЙЙНјааЃЌШєВЛЪЧдкдзЂВсЩъБЈЪБЕФМьбщЛњЙЙНјааЃЌгІЬсНЛаТдіаЭКХЕФЕчДХМцШнадШЋЯюФПМьбщБЈИцЃЌДцдкЖрИіаТдіаЭКХЃЌгІГіОпЛљгкаТдіаЭКХЕФИВИЧвтМћЁЃ
ЁЁЁЁ(2)аТдіМгВњЦЗаЭКХЃЌдаЭКХЭЌЪБЗЂЩњБфЛЏ
ЁЁЁЁгІЖдШЋВПаЭКХЕФБфЛЏзїећЬхЦРЙРЃЌГіОпВювьВПЗжЕФВЙГфМьбщБЈИцЃЌВЂгыЪзДЮзЂВсЩъБЈЪБЕФЕчДХМцШнадБЈИцећЬхЙиСЊЃЌгІдкММЪѕвЊЧѓдЄЦРМлБэЛђВЙГфМьбщБЈИцБИзЂРИжаЬхЯжЁЃБфЛЏВПЗжЕФМьбщддђЩЯгІдкдМьбщЛњЙЙНјааЃЌШєВЛЪЧдкдМьбщЛњЙЙНјааЃЌгІећЬхЦРЙРКѓЬсНЛШЋВПаЭКХЕФЕчДХМцШнадМьбщБЈИцЃЌВЂГіОпИВИЧвтМћЃЌМьбщЛњЙЙвВПЩбЁдёЖддзЂВсМьбщБЈИцЦРЙРКѓНјааШЋВПаЭКХЕФВювьЛЏВЙГфМьбщЃЌВЂГіОпИВИЧвтМћЁЃ
ЁЁЁЁ(3)ЮоаТдіМгВњЦЗаЭКХЃЌНідзЂВсаЭКХЗЂЩњБфЛЏ
ЁЁЁЁгІГіОпБфЛЏВПЗжЕФВЙГфМьбщБЈИцЃЌВЂгыЪзДЮзЂВсЩъБЈЪБЕФЕчДХМцШнадБЈИцЙиСЊЃЌгІдкММЪѕвЊЧѓдЄЦРМлБэЛђВЙГфМьбщБЈИцБИзЂРИжаЬхЯжЁЃБфЛЏВПЗжЕФМьбщддђЩЯгІдкдМьбщЛњЙЙНјааЃЌШєВЛЪЧдкдзЂВсЩъБЈЪБЕФМьбщЛњЙЙНјааЃЌгІЬсНЛБфЛЏаЭКХЕФШЋЯюФПЕчДХМцШнадМьбщБЈИцЃЌЖрИіаЭКХБфЛЏЪБЃЌЛЙгІГіОпаЭКХИВИЧвтМћЃЌМьбщЛњЙЙвВПЩбЁдёЖддзЂВсМьбщБЈИцЦРЙРКѓНјааБфЛЏаЭКХЕФВювьЛЏВЙГфМьбщЃЌВЂГіОпИВИЧвтМћЁЃ
ЁЁЁЁ(4)ЖдвдЩЯБфЛЏЧщПіЃЌМьбщЛњЙЙЛЙгІЖдВњЦЗЕчДХМцШнадв§Ц№ЕФАВЙцБфЛЏзїЙиСЊадЦРЙРЃЌБивЊЪБЃЌГіОпАВЙцЕФВювьЛЏВЙГфМьбщБЈИцЃЌгІдкММЪѕвЊЧѓдЄЦРМлБэЛђВЙГфМьбщБЈИцБИзЂРИжаЬхЯжећЬхЙиСЊадЁЃ
ЁЁЁЁ(Ш§)вНСЦЦїаЕбгајзЂВс
ЁЁЁЁбгајзЂВсЪБЃЌВњЦЗддђЩЯВЛгІЗЂЩњзЂВсжЄМАЦфИНМўдиУїЪТЯюЕФБфЛЏЁЃЕЋПЩФмДцдквЛаЉЬиЪтЧщаЮЕФБфЛЏЃЌжївЊАќРЈЃКЧщаЮвЛЃЌзЂВсШЫЮЊЪЪгІЙњМвЧПжЦадБъзМБфЛЏЁЂЙњМвБъзМЦЗЁЂВЮПМЦЗЗЂВМЛђИќаТЪЕЪЉЃЌаоИФВњЦЗММЪѕвЊЧѓКЭзЂВсжЄдиУїЕФЦфЫћаэПЩЪТЯю(ШчадФмНсЙЙзщГЩЕШ)ЕШЧщаЮ;ЧщаЮЖўЁЂВњЦЗдкЩЯвЛТжзЂВсжмЦкФкЗЂЩњВЛЩцМАаэПЩЪТЯюЕФБфЛЏЁЃ
ЁЁЁЁ1.ВњЦЗУЛгаБфЛЏЩљУї
ЁЁЁЁШєДцдкЩЯЪіБфЛЏЧщаЮЃЌЦѓвЕгІдкВњЦЗУЛгаБфЛЏЩљУїжаЫЕУїБфЛЏФкШнЃЌЬсЙЉЖдБШЫЕУїКЭЯрЙижЇГжзЪСЯЁЃЬсЙЉЕчДХМцШнадУшЪіЮФЕЕЃЌЬхЯжгыВњЦЗЕчДХМцШнадЯрЙиЕФБфЛЏФкШнЃЌгІзЂУїЮДЗЂЩњБфЛЏЕФВПЗжЁЃЖдЧщаЮЖўЕФБфЛЏЃЌзЂВсШЫЛЙгІЬсЙЉ“ВњЦЗЫљЗЂЩњЕФБфЛЏЭЈЙ§жЪСПЙмРэЬхЯЕНјааПижЦЃЌзЂВсжЄдиУїЪТЯюЮоБфЛЏ”ЕФЩљУїЁЃ
ЁЁЁЁ2.ВњЦЗМьбщБЈИц
ЁЁЁЁ(1)ЗЂЩњЧщаЮвЛЕФБфЛЏ
ЁЁЁЁШчЕчДХМцШнЯрЙиЙњМвЧПжЦадБъзМвбаоЖЉЃЌзЂВсШЫгІЬсЙЉВњЦЗЗћКЯЧПжЦадБъзМвЊЧѓЕФМьбщБЈИцЃЌМьбщБЈИцгІгЩОпгазЪжЪЕФМьбщЛњЙЙГіОпЁЃЧПжЦадБъзМБфЛЏАќКЌЦфжав§гУЕФЯожЕКЭМьбщЗНЗЈБъзМЗЂЩњБфЛЏЃЌЬхЭтеяЖЯ(IVD)ЩшБИжагУгкЦРЖЈПЙШХЖШЪдбщНсЙћЕФЙІФмКЭ/ЛђадФмЕФМьбщЗНЗЈЫљЩцМАЕФЯрЙиЙњМвБъзМЦЗЁЂВЮПМЦЗЗЂЩњБфЛЏЁЃМьбщЛњЙЙгІзлКЯЦРЙРВњЦЗЮЊЪЪгІЧПжЦадБъзМЗЂЩњЕФБфЛЏЖдВњЦЗЦфЫћМьбщЯюЕФгАЯьЃЌБивЊЪБЃЌгІГіОпВЙГфМьбщБЈИцЃЌВЂдкМьбщБЈИцБИзЂРИЬхЯжЙиСЊадЁЃШєЧПжЦадБъзМЮоЪЕжЪадБфЛЏЃЌзЂВсШЫПЩЬсЙЉаТОЩБъзМЖдБШБэЃЌЫЕУїЮоЪЕжЪадБфЛЏЕФвРОнЁЃ
ЁЁЁЁ(2)ЗЂЩњЧщаЮЖўЕФБфЛЏ
ЁЁЁЁИљОнЬсЙЉЕФЕчДХМцШнадУшЪіЮФЕЕЃЌШєБфЛЏВПЗжЖдВњЦЗЕчДХМцШнадДцдкгАЯьЃЌНЈвщЬсЙЉОпгазЪжЪЕФМьбщЛњЙЙГіОпЕФЦРЙРБЈИцзїЮЊБфЛЏВЛгАЯьВњЦЗЗћКЯадЕФжЇГжзЪСЯЃЌвВПЩЬсЙЉЩшМЦПЊЗЂБфИќПижЦЕФбщжЄ/ШЗШЯБЈИцКЭЦРЩѓНсТлЃЌВЂГаХЕБфЛЏКѓЕФВњЦЗФмТњзуВњЦЗММЪѕвЊЧѓЙцЖЈЁЃ
ЁЁЁЁАЫЁЂЩѓВщЙизЂЕу
ЁЁЁЁ(вЛ)зЂВсМьбщЙизЂЕу
ЁЁЁЁ1.ЕчДХМцШнадУшЪіЮФЕЕ
ЁЁЁЁвНСЦЦїаЕзЂВсжЄАьРэШЫЬсЙЉЕФВњЦЗЕчДХМцШнадУшЪіЮФЕЕФкШнЪЧЗёЭъШЋЃЌгыбљЦЗЪЕЮяЪЧЗёвЛжТЁЃ
ЁЁЁЁ(1)ВњЦЗЕчДХМцШнадАВШЋЬиеїЫЕУї
ЁЁЁЁШэМўАцБОКХЭЈГЃгыВњЦЗЕФЙІФм/ЙЄзїФЃЪНУмЧаЯрЙиЁЃВњЦЗдЄЦкгУЭОЁЂдЄЦкЪЙгУЛЗОГМАЪЙгУЯожЦЁЂЪЪгУШЫШКгыВњЦЗЕФЕчДХМцШнЗжзщЗжРрУмЧаЯрЙиЃЌашжиЕуЙизЂЁЃ
ЁЁЁЁЗжзщЗжРраХЯЂжБНггАЯьВњЦЗЕФЕчДХМцШнадМьбщНсТлЃЌашШЗШЯЪЧЗёгыЪЪгУЗЖЮЇЯрЪЪгІЁЃ
ЁЁЁЁЛљБОадФмЕФШЗЖЈгІЗћКЯЦфЖЈвхЃЌгІИљОнIEC 60601МАБОжИФЯЕФЙцЖЈШЗЖЈЃЌБивЊЪБЃЌЛЙгІЬсЙЉЗчЯеЗжЮізЪСЯжаЕчДХМцШнадЯрЙиВПЗжФкШнЁЃжБНгв§гУВњЦЗММЪѕвЊЧѓжаЕФадФмжИБъзїЮЊЛљБОадФмЪБЃЌЦфМьбщЗНЗЈгІгыММЪѕвЊЧѓвЛжТЃЌШєВЛвЛжТЃЌгІЬсЙЉЗНЗЈЦЋРыЖдЪдбщНсЙћЕФгАЯьЗжЮіЁЃ
ЁЁЁЁВњЦЗЙЄзїФЃЪНжБНггАЯьЕчДХМцШнадМьбщНсТлЃЌгІНсКЯбљЦЗЪЕЮяЃЌЩѓВщЩљГЦЕФЙЄзїФЃЪНЪЧЗёОпгаМьбщДњБэадЁЃ
ЁЁЁЁВњЦЗЕФНсЙЙгІгыбљЦЗЪЕЮявЛжТЃЌжиЕуЙизЂВњЦЗгВМўзщГЩВПМў/ФЃПщжЎМфЕФЭтВПСЌНгЙиЯЕЃЌЬсЙЉбЁХфВПМў/ФЃПщЁЂВтЪдгУИЈжњЩшБИЕФаХЯЂгІгыЬсЙЉЕФЪЕЮявЛжТЁЃЯпРТГЄЖШЪЧжИВњЦЗЭтВПЯпРТЕФГЄЖШЃЌБивЊЪБЭЈЙ§ВтСПШЗШЯЃЌЦСБЮЧщПігІгыЪЕЮявЛжТЁЃЙиМќдЊЦїМўЫљСаЦїМўНігыВњЦЗЕчДХМцШнадЯрЙиЃЌВЛЭъШЋЕШЭЌгкВњЦЗАВЙцМьВтЯюФПЩцМАЕФЙиМќдЊЦїМўЃЌвВЭъШЋВЛЕШЭЌгкЦѓвЕжЪСПЙмРэЬхЯЕПижЦжаЕФAРрЮяСЯЃЌЬсЙЉЕФЙиМќдЊЦїМўаХЯЂгІгыЪЕЮяЯрЖдгІЁЃ
ЁЁЁЁКЫЖдЛэУтЪдбщЕФЧщПіЫЕУїЪЧЗёгыбљЦЗЪЕМЪЧщПівЛжТЁЃ
ЁЁЁЁ(2)аЭКХИВИЧЫЕУї
ЁЁЁЁгІЖдЭЌвЛзЂВсЕЅдЊКЌЖрИіаЭКХЕФВњЦЗНјаааЭКХИВИЧЃЌаЭКХИВИЧгІбЁШЁвЛИіЛђЖрИіОпгаМьбщЕфаЭадЕФДњБэаЭКХЃЌжиЕуЙизЂзЂВсШЫЬсЙЉЕФаЭКХИВИЧЫЕУїЪЧЗёЪєЪЕЁЃ
ЁЁЁЁ(3)ГаХЕЪщ
ЁЁЁЁЭЌЪБЫЭМьЭЌвЛаЭКХЖрЬЈбљЦЗНјаааЭЪНМьбщПЩЬсИпМьбщаЇТЪЃЌбљЦЗвЛжТадЪЧЖрЬЈбљЦЗЭЌЪБНјааВЛЭЌЯюФПМьбщЕФГѕЪМЬѕМўЃЌМьбщНсЪјКѓЃЌжївЊГаМьЛњЙЙЛЙгІЦРЙРМьбщЭЈЙ§КѓбљЦЗЕФвЛжТадЁЃ
ЁЁЁЁ2.ВњЦЗММЪѕвЊЧѓКЭМьбщБЈИц
ЁЁЁЁжиЕуЙизЂВњЦЗММЪѕвЊЧѓжаЕФЕчДХМцШнадВЩБъЧщПіЪЧЗёЭъећЃЌАќКЌЕчДХМцШнадЭЈгУБъзМЁЂзЈгУБъзММАЯрЙиВњЦЗжИЕМддђжаЕФЕчДХМцШнадвЊЧѓЕШЁЃ
ЁЁЁЁгІИљОнМьбщЭъГЩКѓЕФбљЦЗАВШЋЬиеїаХЯЂЃЌНЋЙцЖЈФкШнЬхЯждкМьбщБЈИцжаЃЌШчЃКбљЦЗЙЙГЩЁЂСЌНгЭМЁЂЯпРТаХЯЂЁЂЙЄзїФЃЪНЁЂИЈжњЩшБИаХЯЂЁЂЙиМќдЊЦїМўЧхЕЅаХЯЂЕШЁЃ
ЁЁЁЁ3.ВњЦЗЫЕУїЪщ
ЁЁЁЁЫЕУїЪщФкШнЪєгкМьбщЯюФПЃЌгІНсКЯбљЦЗЪЕЮяКЫЖдгыЪЪгУБъзМжаЕФЫЕУїЪщвЊЧѓЃЌжиЕуЙизЂYY 0505жаБэ201жСБэ208ЕФЪЪгУадЁЃ
ЁЁЁЁ(вЛ) ММЪѕЩѓЦРЙизЂЕу
ЁЁЁЁ1.ЕчДХМцШнадУшЪіЮФЕЕ
ЁЁЁЁгІКЫЖдЕчДХМцШнадУшЪіЮФЕЕФкШнгыВњЦЗММЪѕвЊЧѓЁЂЫЕУїЪщЁЂМьбщБЈИцЕФвЛжТадЁЃжиЕуЙизЂбљЦЗдкзЂВсМьбщЙ§ГЬжаЗЂЩњЕФећИФЯюФПЃЌвНСЦЦїаЕзЂВсШЫдкЩъЧызЂВсЪБЃЌгІЬсНЛгызЂВсМьбщећИФЭъГЩКѓЯрЪЪгІЕФВњЦЗЕчДХМцШнадУшЪіЮФЕЕМАЮФЕЕжаЫљЪіФкШнЖдгІЕФжЇГжадзЪСЯЁЃЛЙгІЙизЂећИФЖдВњЦЗЪЙгУЦкЯоКЭЮЌЛЄБЃбјЗНЗЈЕФгАЯьЃЌгІЬсНЛЯрЙиЗжЮіЦРЙРКЭ/ЛђбщжЄзЪСЯЁЃ
ЁЁЁЁ2.ЩњВњжЦдьаХЯЂ
ЁЁЁЁЩцМАВњЦЗКЯЙцадећИФЃЌгІжиЕуЙизЂЩњВњЙЄвеСїГЬЭМКЭЙ§ГЬПижЦЕуЪЧЗёЗЂЩњБфЛЏЁЃШчЃКећИФжаЩцМАЖдВњЦЗФкПЧХчЭПЕМЕчЦсЃЌЖјВњЦЗГѕЪМЩњВњЙЄвеСїГЬЭМКЭЙ§ГЬПижЦЕуЮДАќКЌХчЭПЙЄађМАПижЦЃЌдђгІЬсНЛИќИФКѓЕФЙЄвеЮФМўЁЃШєећИФжаНіИќИФАВзАЕФдЊЦїМўЃЌЪєгкГѕЪМЩњВњЙЄвежаЕФзщзАЙЄађЃЌЮДИФБфГѕЪМЙЄвеЃЌдђВЛЩцМАЩњВњЙЄвеСїГЬЭМКЭЙ§ГЬПижЦЕуЕФИФБфЁЃ
ЁЁЁЁ3.ВњЦЗЗчЯеЗжЮізЪСЯ
ЁЁЁЁвНСЦЦїаЕзЩбЏЬсабФњгІИљОнIEC 60601МАБОжИФЯЕФЙцЖЈЃЌНсКЯЗчЯеЗжЮізЪСЯЃЌЩѓВщЛљБОадФм/ЦРЖЈПЙШХЖШЪдбщНсЙћЕФадФмКЭ/ЛђЙІФмЁЂЗћКЯадзМдђ/адФмХаОнЪЧЗёКЯРэЁЃ
ЁЁЁЁ4.ВњЦЗММЪѕвЊЧѓ
ЁЁЁЁжиЕуЩѓВщМьбщЛњЙЙГіОпЕФДњБэадВњЦЗбЁШЁМАаЭКХИВИЧЕФНсТлвтМћЃЌЪЧЗёКИЧСЫШЋВПЩъБЈаЭКХЁЃЩѓВщдЄЦРМлвтМћЕФМьбщБЈИцБрКХЁЂЪЪгУБъзМКХЁЂИВИЧЫЕУїЕШЪЧЗёЭъШЋЁЃ
ЁЁЁЁ5.ВњЦЗзЂВсМьбщБЈИц
ЁЁЁЁАДееЁЖЙњМвЪГЦЗвЉЦЗМрЖНЙмРэОжАьЙЋЬќЙигкгЁЗЂYY 0505МьбщБЈИцВЮПМИёЪНЕФЭЈжЊЁЗ(ЪГвЉМрАьаЕЙм[2013]29КХ)ЃЌжиЕуКЫВщМьбщБЈИцЕФЪмМьбљЦЗаХЯЂЁЂбљЦЗЙЙГЩЁЂбљЦЗСЌНгЭМЁЂбљЦЗдЫааФЃЪНЁЂбљЦЗЕчРТЁЂИЈжњЩшБИЁЂЪдбщНсЙћИХЪіЪЧЗёгыЕчДХМцШнадУшЪіЮФЕЕвЛжТЃЌЪдбщвЊЧѓКЭЪ§ОнжаЕФЪдбщУшЪіЪЧЗёгыЬсЙЉЕФЛэУтЧщПівЛжТЃЌееЦЌвГЕФбљЦЗЪЕЮяЭМЦЌЪЧЗёЬхЯжСЫећИФаХЯЂЃЌЙиМќдЊЦїМўЧхЕЅЪЧЗёгыЬсЙЉЕФвЛжТЁЃ
ЁЁЁЁ6.ВњЦЗЫЕУїЪщ
ЁЁЁЁжиЕуЩѓВщВњЦЗЫЕУїЪщжаЕФЕчДХМцШнадаХЯЂгыВњЦЗЪЪгУЗЖЮЇЕФЪЪгІадЁЃ
ЁЁЁЁ7.ЗћКЯадЩљУї
ЁЁЁЁЩѓВщВњЦЗашЗћКЯЕФЕчДХМцШнадЭЈгУБъзМЁЂзЈгУБъзМЁЂВњЦЗжИЕМддђЪЧЗёАќКЌЭъШЋЁЃ
ЁЁЁЁ8.аэПЩЪТЯюБфИќ
ЁЁЁЁжиЕуЩѓВщВњЦЗаЭКХБфЛЏЕФИїжжЧщПіЃЌЪЧЗёЬсЙЉБфЛЏВПЗжЕФЕчДХМцШнадМьбщБЈИцЁЂЙиСЊадЫЕУїЁЂИВИЧвтМћЁЃ
ЁЁЁЁ9.бгајзЂВс
ЁЁЁЁжиЕуЩѓВщВњЦЗдкбгајЪБЗЂЩњЕФЪЪгІЕчДХМцШнЧПжЦадБъзМБфЛЏКЭВЛЩцМАаэПЩЪТЯюЕФБфЛЏЃЌЪЧЗёЬсНЛЯргІЕФМьбщБЈИцЛђЩшМЦПЊЗЂБфИќПижЦЕФЦРЩѓзЪСЯЁЃ
ЁЁЁЁОХЁЂВЮПМЮФЯз
ЁЁЁЁ[1]ЁЖвНСЦЦїаЕзЂВсЙмРэАьЗЈЁЗ(ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжСюЕк4КХ)
ЁЁЁЁ[2]ЁЖвНСЦЦїаЕЫЕУїЪщКЭБъЧЉЙмРэЙцЖЈЁЗ(ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжСюЕк6КХ)
ЁЁЁЁ[3] ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙигкЗЂВМвНСЦЦїаЕВњЦЗММЪѕвЊЧѓБраДжИЕМддђЕФЭЈИц(ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЭЈИц2014ФъЕк9КХ)
ЁЁЁЁ[4] ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙигкЙЋВМвНСЦЦїаЕзЂВсЩъБЈзЪСЯвЊЧѓКЭХњзМжЄУїЮФМўИёЪНЕФЙЋИц(ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙЋИц2014ФъЕк43КХ)
ЁЁЁЁ[5] ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙигкЪЕЪЉЁЖвНСЦЦїаЕзЂВсЙмРэАьЗЈЁЗКЭЁЖЬхЭтеяЖЯЪдМСзЂВсЙмРэАьЗЈЁЗгаЙиЪТЯюЕФЭЈжЊ(ЪГвЉМраЕЙм[2014]144КХ)
ЁЁЁЁ[6] ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙигкЗЂВМвНСЦЦїаЕСйДВЦРМлММЪѕжИЕМддђЕФЭЈИц(ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЭЈИц2015ФъЕк14КХ)
ЁЁЁЁ[7] ЪГЦЗвЉЦЗМрЙмзмОжЙигкжДаавНСЦЦїаЕКЭЬхЭтеяЖЯЪдМСзЂВсЙмРэАьЗЈгаЙиЮЪЬтЕФЭЈжЊ(ЪГвЉМраЕЙмЁВ2015ЁГ247КХ)
ЁЁЁЁ[8] YY 0505-2012вНСЦЦїаЕаавЕБъзМЪЕЪЉЙЄзїЗНАИ(ЪГвЉМрАьаЕ[2012]149КХ)
ЁЁЁЁ[9] ЙигкYY 0505-2012вНСЦЦїаЕаавЕБъзМЪЕЪЉгаЙиЙЄзївЊЧѓЕФЭЈжЊ(ЪГвЉМрАьаЕ[2012]151КХ)
ЁЁЁЁ[10] ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэОжАьЙЋЬќЙигкгЁЗЂYY 0505МьбщБЈИцВЮПМИёЪНЕФЭЈжЊ(ЪГвЉМрАьаЕЙм[2013]29КХ)
ЁЁЁЁ[11]ЙњМвЪГЦЗвЉЦЗМрЖНЙмРэзмОжЙигкЗЂВМвНСЦЦїаЕЩњВњжЪСПЙмРэЙцЗЖЕФЙЋИц(2014ФъЕк64КХ)
ЁЁЁЁ[12] вНСЦЦїаЕзЂВсЕЅдЊЛЎЗжжИЕМддђ
ЁЁЁЁ[13] вНСЦЦїаЕШэМўзЂВсММЪѕЩѓВщжИЕМддђ
ЁЁЁЁ[14] ЙЧзщжЏЪжЪѕЩшБИзЂВсММЪѕЩѓВщжИЕМддђ(2017ФъаоЖЉАц)
ЁЁЁЁ[15] GB 4343.1-2009ЁЖМвгУЕчЦїЁЂЕчЖЏЙЄОпКЭРрЫЦЦїОпЕФЕчДХМцШнвЊЧѓЁЁЕк1ВПЗжЃКЗЂЩфЁЗ
ЁЁЁЁ[16] GB 9254-2008ЁЖаХЯЂММЪѕЩшБИЕФЮоЯпЕчЩЇШХЯожЕКЭВтСПЗНЗЈЁЗ
ЁЁЁЁ[17] GB 4824-2019ЁЖЙЄвЕЁЂПЦбЇКЭвНСЦЩшБИ ЩфЦЕЩЇШХЬиад ЯожЕКЭВтСПЗНЗЈЁЗ
ЁЁЁЁ[18] GB 9706.1-2020ЁЖвНгУЕчЦјЩшБИ Ек1ВПЗжЃКЛљБОАВШЋКЭЛљБОадФмЕФЭЈгУвЊЧѓЁЗ
ЁЁЁЁ[19] GB 17625.1-2012ЁЖЕчДХМцШн ЯожЕ аГВЈЕчСїЗЂЩфЯожЕ(ЩшБИУПЯрЪфШыЕчСї≤16A)ЁЗ
ЁЁЁЁ[20] GB 17625.2-2007ЁЖЕчДХМцШн ЯожЕ ЖдУПЯрЖюЖЈЕчСї≤16AЧвЮоЬѕМўНгШыЕФЩшБИдкЙЋЙВЕЭбЙЙЉЕчЯЕЭГжаВњЩњЕФЕчбЙБфЛЏЁЂЕчбЙВЈЖЏКЭЩСЫИЕФЯожЦЁЗ
ЁЁЁЁ[21] GB/T 17625.7-2013ЁЖЕчДХМцШн ЯожЕ ЖдЖюЖЈЕчСї≤75AЧвгаЬѕМўНгШыЕФЩшБИдкЙЋгУЕчбЙЙЉЕчЯЕЭГжаВњЩњЕФЕчбЙБфЛЏЁЂЕчбЙВЈЖЏКЭЩСЫИЕФЯожЦЁЗ
ЁЁЁЁ[22] GB/T17625.8—2015ЁЖЕчДХМцШн ЯожЕ УПЯрЪфШыЕчСїДѓгк16A аЁгкЕШгк75A СЌНгЕНЙЋгУЕЭбЙЯЕЭГЕФЩшБИВњЩњЕФаГВЈЕчСїЯожЕЁЗ
ЁЁЁЁ[23] GB/T 17626.2-2018ЁЖЕчДХМцШн ЪдбщКЭВтСПММЪѕ ОВЕчЗХЕчПЙШХЖШЪдбщЁЗ
ЁЁЁЁ[24] GB/T 17626.3-2016ЁЖЕчДХМцШн ЪдбщКЭВтСПММЪѕ ЩфЦЕЕчДХГЁЗјЩфПЙШХЖШЪдбщЁЗ
ЁЁЁЁ[25] GB/T 17626.4-2018ЁЖЕчДХМцШн ЪдбщКЭВтСПММЪѕ ЕчПьЫйЫВБфТіГхШКПЙШХЖШЪдбщЁЗ
ЁЁЁЁ[26] GB/T 17626.5-2019ЁЖЕчДХМцШн ЪдбщКЭВтСПММЪѕ РЫгП(ГхЛї)ПЙШХЖШЪдбщЁЗ
ЁЁЁЁ[27] GB/T 17626.6-2017ЁЖЕчДХМцШн ЪдбщКЭВтСПММЪѕ ЩфЦЕГЁИагІЕФДЋЕМЩЇШХПЙШХЖШЁЗ
ЁЁЁЁ[28] GB/T 17626.8-2006ЁЖЕчДХМцШн ЪдбщКЭВтСПММЪѕ ЙЄЦЕДХГЁПЙШХЖШЪдбщЁЗ
ЁЁЁЁ[29] GB/T 17626.11-2006ЁЖЕчДХМцШн ЪдбщКЭВтСПММЪѕ ЕчбЙднНЕЁЂЖЬЪБжаЖЯКЭЕчбЙБфЛЏЕФПЙШХЖШЪдбщЁЗ
ЁЁЁЁ[30] GB/T 17743ЁЖЕчЦјееУїКЭРрЫЦЩшБИЕФЮоЯпЕчЩЇШХЬиадЕФЯожЕКЭВтСПЗНЗЈЁЗ
ЁЁЁЁ[31] GB/T 18268.1-2010ЁЖВтСПЁЂПижЦКЭЪЕбщЪвгУЕФЕчЩшБИ ЕчДХМцШнадвЊЧѓ Ек1ВПЗжЃКЭЈгУвЊЧѓЁЗ
ЁЁЁЁ[32] GB/T 18268.26ЁЖВтСПЁЂПижЦКЭЪЕбщЪвгУЕФЕчЩшБИ ЕчДХМцШнадвЊЧѓ Ек26ВПЗжЃКЬхЭтеяЖЯ(IVD)вНСЦЩшБИЁЗ
ЁЁЁЁ[33] YY/T 0316-2016ЁЖвНСЦЦїаЕ ЗчЯеЙмРэЖдвНСЦЦїаЕЕФгІгУЁЗ
ЁЁЁЁ[34] YY 0505-2012ЁЖвНгУЕчЦјЩшБИ Ек1-2ВПЗжЃКАВШЋЭЈгУвЊЧѓ ВЂСаБъзМЃКЕчДХМцШн вЊЧѓКЭЪдбщЁЗБъзМНтЖС
ЁЁЁЁ[35] IEC 60601-1:2012,ЁЖMedical electrical equipment—Part1ЃКGeneral requirements for basic safety and essential performanceЁЗ
ЩюлкКшдЖвНСЦЦїаЕзЩбЏгаЯоЙЋЫОЪЧвЛМвММЪѕзЈвЕЕФвНСЦЦїаЕзЩбЏЙЋЫОЃЌзЈзЂЬсЙЉШЋЙњИїЕиШчЃКЩюлкЁЂЙужнЁЂЖЋнИЁЂжаЩНЁЂЗ№ЩНЁЂГБжнЁЂЫГЕТЁЂЩЯКЃЁЂЮїАВЁЂжиЧьЁЂГЩЖМЕШжЊУћГЧЪаЕФвНСЦЦїаЕСьгђММЪѕзЩбЏЗўЮёЁЃКшдЖвНСЦЦїаЕзЩбЏзЈвЕЗўЮёгкЃКвНСЦЦїаЕВњЦЗзЂВсжЄАьРэЁЂвНСЦЦїаЕЩњВњаэПЩжЄЁЂНјПквНСЦЦїаЕзЂВсЁЂЬхЭтеяЖЯЪдМСзЂВсДњАьРэзЩбЏЁЂвЛРрвНСЦЦїаЕВњЦЗБИАИМАЩњВњБИАИДњАьЁЂвНСЦЦїаЕОгЊаэПЩжЄДњАьЁЂЖўРрвНСЦЦїаЕОгЊБИАИЁЂвНСЦЦїаЕЗжРрНчЖЈЁЂCEШЯжЄЁЂISO13485ШЯжЄЁЂFDAзЂВсЁЂСйДВЪдбщЁЂвНСЦЦїаЕжЪСПЙмРэЬхЯЕШЯжЄМАЬхЯЕНЈСЂгыЙ§ГЬШЗШЯЮФМўНЈСЂ(ISO9001, ISO13485, GMP, CEЃЌQSR820ЃЌCMDCAS);ВњЦЗММЪѕвЊЧѓжЦЖЉЁЂММЪѕЮФМўЁЂСйДВЪдбщМАУтСйДВЭЌРрВњЦЗБШЖдБШзЪСЯБраДЁЂзЂВсзЪСЯБраДИЈЕМЁЂЕчДХМцШндЄВтећИФЁЂвНСЦЦїаЕЙуИцХњЮФЩъБЈЁЂвНСЦЦїаЕГіПкЯњЪлжЄУїАьРэЁЂНрОЛЪвНЈЩшжИЕМЕШЬсЙЉвЛеОЪНЗўЮёЃЌММЪѕзЈвЕЃЌГЯаХЗўЮёЃЌЛЖгФњзЩбЏЃЁ
ЁЁЁЁ
|