|
ЎЎЎЎ
ЎЎЎЎТҪБЖЖчРөЧўІбДЪҝъҫөКЦКх¶ҜБҰЙиұёІъЖ·ЧўІбЦӨ°мАнёщҫЭЎ¶ДЪҝъҫөКЦКх¶ҜБҰЙиұёЧўІбЙуІйЦёөјФӯФтЎ·Ј¬РиТӘМбҪ»өДЙкұЁЧКБПТӘЗуИзПВЈә
ЎЎЎЎ(Т»)ја№ЬРЕПў
ЎЎЎЎ1.ІъЖ·ГыіЖ
ЎЎЎЎІъЖ·өДГьГыУҰ·ыәПЎ¶ТҪБЖЖчРөНЁУГГыіЖГьГы№жФтЎ·(Фӯ№ъјТКіЖ·Т©Ж·ја¶Ҫ№ЬАнЧЬҫЦБоөЪ19әЕ)әН№ъјТұкЧјЎўРРТөұкЧјЦРөДНЁУГГыіЖТӘЗуЎЈІъЖ·ГыіЖУЙТ»ёцәЛРДҙКәНІ»і¬№эИэёцөДМШХчҙКЧйіЙЎЈИфІъЖ·№ӨҫЯН·ҪцОӘЕЩПчН·Ј¬ФтГьГыОӘДЪҝъҫөКЦКхЕЩПчЙиұёЎЈИфІъЖ·ә¬УРіэЕЩПчН·ТФНвөДЖдЛы№ӨҫЯН·(ИзДҘН·өИУГУЪ№ЗЧйЦҜЗРіэөД№ӨҫЯН·)Ј¬ФтГьГыОӘДЪҝъҫөКЦКх¶ҜБҰЙиұёЎЈ
ЎЎЎЎ2.·ЦАаұаВл
ЎЎЎЎДЪҝъҫөКЦКх¶ҜБҰЙиұёЈ¬°ҙХХЎ¶ТҪБЖЖчРө·ЦАаДҝВјЎ·(Фӯ№ъјТКіЖ·Т©Ж·ја¶Ҫ№ЬАнЧЬҫЦ№«ёж2017ДкөЪ104әЕ)Ј¬·ЦАаұаВлОӘ01-09-01Ј¬°ҙ№ъјТТ©Ж·ја¶Ҫ№ЬАнҫЦ2020ДкөЪ147әЕОД,АаұрөчХыОӘ°ҙөЪ¶юАаТҪБЖЖчРө№ЬАнЎЈ
ЎЎЎЎ3.ЧўІбөҘФӘ»®·Ц
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІбөҘФӘ»®·ЦУҰ·ыәПЎ¶ТҪБЖЖчРөЧўІбөҘФӘ»®·ЦЦёөјФӯФтЎ·(Фӯ№ъјТКіЖ·Т©Ж·ја¶Ҫ№ЬАнЧЬҫЦНЁёж2017ДкөЪ187әЕ)өДТӘЗуЈ¬ФӯФтЙПТФІъЖ·өДјјКхФӯАнЎўҪб№№ЧйіЙЎўРФДЬЦёұкәНККУГ·¶О§ОӘ»®·ЦТАҫЭЎЈДЪҝъҫөКЦКх¶ҜБҰЙиұёЧўІбөҘФӘ»®·ЦҪЁТйЦШөг№ШЧўТФПВјёёц·ҪГжЈә
ЎЎЎЎ(1)Ҫб№№ЧйіЙ
ЎЎЎЎІ»Н¬Ҫб№№өДІъЖ·Ј¬ТЛ»®·ЦОӘІ»Н¬өДЧўІбөҘФӘЎЈ
ЎЎЎЎИзЈәЙиұё№©өз·ҪКҪІ»Н¬Ј¬ИзНшөзФҙ№©өзәНДЪІҝөзФҙ№©өзЈ¬ТЛ»®·ЦОӘІ»Н¬өДЧўІбөҘФӘЎЈ¶ФУЪЦч»ъЦРІ»ә¬өз»ъөДІъЖ·Ј¬ИфКЦұъөҘ¶АЧўІбЈ¬Т»МеКҪКЦұъәН·ЦМеКҪКЦұъөДЙиұёТЛ»®·ЦОӘІ»Н¬ЧўІбөҘФӘЎЈ
ЎЎЎЎ№ӨҫЯН·ҝЙУлЦч»ъТ»Н¬ЧўІбЈ¬ТІҝЙөҘ¶АЧўІбЎЈ
ЎЎЎЎ(2)ККУГ·¶О§
ЎЎЎЎІ»Н¬ІъЖ·ИфККУГ·¶О§І»Н¬Ј¬ФӯФтЙП»®·ЦОӘІ»Н¬өДЧўІбөҘФӘЎЈ
ЎЎЎЎИзЈәҪцУГУЪұЗЗ»ІҝО»УлҪцУГУЪ°тлЧІҝО»өДДЪҝъҫөКЦКхЕЩПчЙиұё»®·ЦОӘІ»Н¬ЧўІбөҘФӘЎЈ
ЎЎЎЎЧўЈәТ»ёцІъЖ·УР¶аёцІҝО»өДУҰУГЈ¬І»Йжј°ЧўІбөҘФӘ»®·ЦЎЈ
ЎЎЎЎ4.ТҪБЖЖчРө°ІИ«әНРФДЬ»щұҫФӯФт
ЎЎЎЎТҪБЖЖчРөЧўІб°мАнЙкЗлИЛҝЙёщҫЭІъЖ·МШРФЕР¶ПТҪБЖЖчРө°ІИ«РФДЬ»щұҫФӯФтЦРёчМхҝоөДККУГРФЈ¬ЦӨГч·ыәПРФІЙУГөД·Ҫ·ЁәНОӘ·ыәПРФМṩҝН№ЫЦӨҫЭөДОДјюУЙЙкЗлИЛёщҫЭКөјКЗйҝцҪшРРМоРҙЎЈОӘ·ыәПРФМṩөДЦӨҫЭИз°ьә¬ФЪІъЖ·ЧўІбЙкұЁЧКБПЦРЈ¬УҰөұФЪОДјюЦРЛөГчЖдФЪЙкұЁЧКБПЦРөДҫЯМеО»ЦГЎЈАэИзЈәЧўІбјмСйұЁёж(ТҪУГөзЖш°ІИ«Јә»ъРө·зПХөД·А»ӨІҝ·Ц);ЛөГчКйөЪ4.2ХВЎЈ¶ФУЪОҙ°ьә¬ФЪТҪБЖЖчРөІъЖ·ЧўІбЙкұЁЧКБПЦРөДОДјюЈ¬УҰөұЧўГчёГЦӨҫЭОДјюГыіЖј°ЖдФЪЦКБҝ№ЬАнМеПөОДјюЦРөДұаәЕұёІйЎЈ¶ФУЪІ»ККУГөДМхҝоЈ¬УҰҪбәПІъЖ·МШөгЛөГчАнУЙЎЈ
ЎЎЎЎ(¶ю)ЧЫКцЧКБП
ЎЎЎЎТҪБЖЖчРөЧўІбЦӨЙкЗлУҰГиКцІъЖ·ЦчТӘ№ҰДЬЎўКөПЦФӨЖЪУГНҫөД·Ҫ·ЁТФј°ЗшұрУЪН¬АаІъЖ·өДМШХчЎЈјјКхЙуЖАЦШөг№ШЧўІъЖ·өДҪб№№ЎўІДБПЎўК№УГ·Ҫ·ЁЎўРФДЬІОКэј°ЖдУлТСЙПКРІъЖ·өДІоТмЎЈТФј°ІоТмІъЙъөДФӯТтЈ¬ұИИзЈ¬КЗ·сёДҪшБЛЙијЖёь·ыәПБЩҙІөДРиЗуөИЎЈ
ЎЎЎЎёГАаІъЖ·РиЦШөг№ШЧўЧӘЛЩ»тНщёҙЖөВКТФј°ОьТэБчЛЩЎЈ
ЎЎЎЎ1.№ӨЧчФӯАн
ЎЎЎЎёГАаІъЖ·РиЕдәПДЪҝъҫөК№УГЈ¬УЙөзФҙЗэ¶Ҝ¶ҜБҰЧ°ЦГЈ¬¶ҜБҰЧ°ЦГОӘ№ӨҫЯН·Мṩ»ъРө¶ҜБҰЗэ¶Ҝ№ӨҫЯН·РэЧӘ»тНщёҙФЛ¶ҜЎЈ
ЎЎЎЎІъЖ·ХэіЈ№ӨЧчКұЈ¬№ӨҫЯН·ҪУҝЪІҝО»ІеИлКЦұъҪУҝЪІҝО»Ј¬КЦұъФЪЦч»ъәН/»тҪЕМӨҝӘ№Ш(ИфУР)өДҝШЦЖПВКдіцөҘПт»тНщёҙөДРэЧӘФЛ¶ҜЈ¬ҙш¶Ҝ№ӨҫЯН·РэЧӘІҝ·ЦРэЧӘЈ¬КөК©¶ФИЛМеЧйЦҜ»тТмОпөДҪКЛйәНЗРіэЎЈК№УГ№эіМЦРЈ¬№ӨҫЯН·ХыМеҙҰУЪідВъөИЙшТәөДТәМе»·ҫіЦРЈ¬өИЙшТәҝЙҫӯ№ӨҫЯН·НЁөАЧўИлЈ¬»тХЯУЙЖдЛьЖчРөөҘ¶АЧўИлЈ¬И»әуөИЙшТәУлұ»ЗРПчПВАҙөДЧйЦҜТ»ЖрФЪёәС№ЧчУГПВҙУ№ӨҫЯН·әНКЦұъөДЦРҝХНЁөАЕЕіцЎЈұЗсјУлВӯөЧөДУҰУГФт¶аОӘЖшМе»·ҫіЎЈёәС№Ч°ЦГҝЙУЙ¶АБўөДөз¶ҜОьТэЖчМṩЈ¬»тХЯУЙІъЖ·ЧФҙшёәС№Ч°ЦГМṩЎЈ
ЎЎЎЎКЦКхЦРЕдәПК№УГөДДЪҝъҫө¶аОӘУІРФДЪҝъҫөЈ¬Из№ШҪЪДЪҝъҫөЎўұЗсјДЪҝъҫөөИЎЈ
ЎЎЎЎ№ӨҫЯН·НЁіЈҫӯУлДЪҝъҫөПаН¬өД»тІ»Н¬өДНЁөАҪшИлИЛМеЎЈНЁөА·ЦОӘҙҙҝЪНЁөАәНИЛМеЧФИ»З»өАЎЈ
ЎЎЎЎ2.Ҫб№№ЧйіЙ
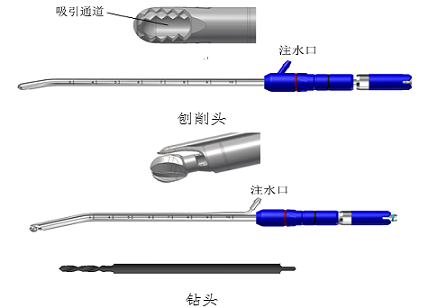
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІбЦӨЙкЗлёГАаІъЖ·НЁіЈУЙЦч»ъЎўКЦұъЎў№ӨҫЯН·(ЕЩПчН·ЎўДҘН·ЎўЧкН·өИ)ЎўҪЕМӨҝӘ№Ш(ИфУР)ЎўёҪјюЧйіЙЎЈ
ЎЎЎЎ(1)Цч»ъОӘКЦұъМṩөзДЬ»т»ъРөДЬЈ¬Іў¶ФЖдКдіцКөК©јаҝШЎЈИфІъЖ·ЧФҙшёәС№ОьТэ№ҰДЬЈ¬Цч»ъ»№ҝЙОӘёГ№ҰДЬМṩёәС№ЎЈ
ЎЎЎЎ(2)КЦұъУЙІЩЧчХЯОХіЦІўЗэ¶ҜЛщјРіЦөД№ӨҫЯН·КөПЦКЦКхЎЈКЦұъҝЙОӘТ»МеКҪ»т·ЦМеКҪЎЈТ»МеКҪКЦұъНЁіЈДЪЦГөз»ъІўјҜіЙҙ«¶ҜәН№ӨҫЯН·ҪУҝЪІҝ·ЦЎЈ·ЦМеКҪКЦұъУЙјРіЦ№ӨҫЯН·өДКЦұъәНёшКЦұъМṩ¶ҜБҰөДөз»ъБҪІҝ·ЦЧйіЙЎЈІҝ·ЦІъЖ·өДөз»ъІ»ФЪКЦұъЦР¶шДЪЦГФЪЦч»ъЦРЎЈІҝ·ЦКЦұъҙшУРёәС№ОьТэНЁөАЎЈ
ЎЎЎЎ(3)№ӨҫЯН·јРіЦФЪКЦұъЙПЈ¬¶ФИЛМеЧйЦҜ»тТмОпКөК©ҪКЛйЎўЗРіэЎЈ№ӨҫЯН·ЦЦАаҪП¶аЈ¬ҪПОӘіЈјыөДУРЕЩПчН·ЎўДҘН·ЎўЧкН·өИЎЈ
ЎЎЎЎЕЩПчН·УЙДЪРэЧӘІҝ·ЦУлҙшҙ°ҝЪөДНв№М¶ЁІҝ·Ц№№іЙЎЈДЪРэЧӘІҝ·ЦУЙДЪө¶Н·ЎўДЪө¶№ЬЎўДЪө¶ҪУҝЪЧйіЙЈ¬ҙшҙ°ҝЪНв№М¶ЁІҝ·ЦУЙНвө¶Н·/»ӨЗК(ИфУР)ЎўНвө¶№ЬЎўНвө¶ҪУН·ЧйіЙЎЈЕЩПчН·РиҪ«ИнЧйЦҜОьИлөҪДЪРэЧӘІҝ·ЦЈ¬Тт¶ш№ӨҫЯН·ДЪРэЧӘІҝ·Цә¬УРОьТэНЁөАЎЈ
ЎЎЎЎДҘН·НЁіЈ°ьә¬РэЧӘІҝ·ЦЈ¬ЙЩКэДҘН·УЙРэЧӘІҝ·ЦУлҙшҙ°ҝЪөДНв№М¶ЁІҝ·Ц№№іЙЎЈРэЧӘІҝ·ЦУЙН·¶Л№ӨЧчІҝ·ЦЎўұъІҝЎўОІІҝҪУҝЪЧйіЙЈ¬ҙшУРНв№М¶ЁІҝ·ЦУЙ»ӨЗКЎўНв№ЬЎўНв№ЬҪУН·ЧйіЙЎЈҙшУРНв№М¶ЁІҝ·ЦҪб№№өДДҘН·ә¬УРОьТэНЁөАЎЈ
ЎЎЎЎЧкН·НЁіЈҪц°ьә¬РэЧӘІҝ·ЦЈ¬РэЧӘІҝ·ЦУЙН·¶Л№ӨЧчІҝ·ЦЎўұъІҝәНОІІҝҪУҝЪЧйіЙЎЈ
ЎЎЎЎ(4)ҪЕМӨҝӘ№ШНЁіЈ°ҙУлЦч»ъөДБ¬ҪУ·ҪКҪ·ЦОӘөзАВБ¬ҪУәНОЮПЯБ¬ҪУЎЈЎЈ
ЎЎЎЎ(5)УлЙиұёҙжФЪөзЖш»тОпАнБ¬ҪУөДёҪјюЈ¬ЦчТӘ°ьАЁөзФҙПЯЎўёәС№өчҪЪЖчЎўКХјҜИЭЖчөИЎЈ
ЎЎЎЎИфЧйіЙЦРә¬¶аёцРНәЕЈ¬ЧЫКцЧКБПЦРУҰГчИ·ёчРНәЕЦ®јдөДІоТмЈ¬ұШТӘКұМṩͼКҫЎЈІъЖ·јјКхТӘЗуј°ЛөГчКйЦРТІУҰёщҫЭІъЖ·ҫЯМеЗйҝцГчИ·ұҫЧўІбөҘФӘДЪёчРНәЕ/№жёсІъЖ·өДҪб№№әНЧйіЙЎЈ
ЎЎЎЎ3.ТҪБЖЖчРөЧўІбЦӨЙкЗлІъЖ·ККУГ·¶О§әНҪыјЙЦў
ЎЎЎЎёГАаІъЖ·НЁіЈУГУЪЧйЦҜөДҪКЛйәНЗРіэЎЈККУГ·¶О§өДұнКцУҰёщҫЭ№ӨҫЯН·АаРНЈ¬ГчИ·ККУГөДЧйЦҜАаРНЈ¬»№УҰГчИ·ҝЙЕдәПК№УГДЪҝъҫөөДЦЦАаЎЈАэИзЈәІъЖ·Ул№ШҪЪДЪҝъҫөЕдәПК№УГЈ¬УГУЪҪКЛйәНЗРіэИнЧйЦҜәН№ЗЧйЦҜЎЈІўЧўГч“ФЪТҪБЖ»ъ№№ЦРК№УГ”ЎЈ
ЎЎЎЎёГАаІъЖ·ОЮМШКвҪыјЙЦўЈ¬НЁіЈУлҙоЕдК№УГДЪҝъҫөөДҪыјЙЦўұЈіЦТ»ЦВЎЈ
ЎЎЎЎ4.ІОҝјөДН¬АаІъЖ·»тЗ°ҙъІъЖ·өДЗйҝц
ЎЎЎЎТҪБЖЖчРөЧўІбҙъАнЙкЗлИЛУҰөұМṩͬАаІъЖ·(№ъДЪНвТСЙПКР)»тЗ°ҙъІъЖ·өДРЕПўЈ¬ІыКцЙкЗлЧўІбІъЖ·өДСР·ўұіҫ°әНДҝөДЎЈ¶ФУЪН¬АаІъЖ·Ј¬УҰөұЛөГчСЎФсЖдЧчОӘСР·ўІОҝјөДФӯТтЎЈН¬КұБРұнЛөГчЙкЗлЧўІбІъЖ·УлІОҝјІъЖ·(Н¬АаІъЖ·»тЗ°ҙъІъЖ·)ФЪ№ӨЧчФӯАнЎўҪб№№ЧйіЙЎўРФДЬЦёұкТФј°ФӨЖЪУГНҫөИ·ҪГжөДТмН¬Ј¬ұШТӘКұМṩͼКҫЎЈАэИзЈ¬№ӨҫЯН·өДРОЧҙЎўіЯҙзЈ¬ЧӘЛЩЈ¬ОьТэБчЛЩөИЗшұрЎЈ
ЎЎЎЎ(Иэ)СРҫҝЧКБП
ЎЎЎЎ1.ІъЖ·РФДЬСРҫҝ
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІбЦӨЙкЗлИЛУҰөұМṩІъЖ·РФДЬСРҫҝЧКБПТФј°ІъЖ·јјКхТӘЗуөДСРҫҝәНұаЦЖЛөГчЈ¬°ьАЁ№ҰДЬРФЎў°ІИ«РФЦёұкТФј°УлЦКБҝҝШЦЖПа№ШөДЖдЛыЦёұкөДИ·¶ЁТАҫЭЈ¬ЛщІЙУГөДұкЧј»т·Ҫ·ЁЎўІЙУГөДФӯТтј°АнВЫ»щҙЎЎЈЦёұкөДИ·¶ЁТАҫЭУҰГчИ·ЎўҫЯМеЈ¬І»ДЬБэНіөШГиКцОӘ“ТАҫЭІъЖ·МШөг”Ўў“ТАҫЭБЩҙІРиЗуИ·¶Ё”Ј¬УҰҫЯМеЛөГчІъЖ·МШөгәНБЩҙІРиЗуЎЈЦШөг№ШЧўҝХФШЧӘЛЩЎўёәФШЧӘҫШЎўУІ¶ИЎўОьТэБҝөИІОКэЙи¶ЁөДАнУЙј°ТАҫЭЎЈ
ЎЎЎЎ¶ФУЪІОҝјН¬АаІъЖ·И·¶ЁөДЈ¬УҰМṩͬАаІъЖ·өДПа№ШЧКБПЈ¬АэИзЦчТӘРФДЬІОКэөД¶ФұИЎЈ
ЎЎЎЎ¶ФУЪТАҫЭ№ъјТұкЧјЎўРРТөұкЧјЙи¶ЁөДЦёұкЈ¬УҰ№ШЧўұкЧјЦРКЗ·сёшіцБЛҫЯМеөДТӘЗу(АэИзКэЦөЎўПЮЦө);¶ФУЪОҙёшіцҫЯМеТӘЗуөДЈ¬ЙкЗлИЛУҰЛөГчЙкұЁІъЖ·№ҰДЬЎўРФДЬЦёұкИ·¶ЁөДТАҫЭЈ¬јҙЙијЖКдИлИ·¶ЁөДАнУЙЎЈРФДЬЦёұкҝЙёщҫЭІъЖ·КөјКЗйҝцІОҝјYY/T0955Ў¶ТҪУГДЪҝъҫөДЪҝъҫөКЦКхЙиұёЕЩПчЖчЎ·ЎўYY 0636.1Ў¶ТҪУГОьТэЙиұёөЪ1Іҝ·ЦЈәөз¶ҜОьТэЙиұё°ІИ«ТӘЗуЎ·ЎўYY 0636.3Ў¶ТҪУГОьТэЙиұё өЪ3Іҝ·ЦЈәТФёәС№»тС№БҰФҙОӘ¶ҜБҰөДОьТэЙиұёЎ·ЎўYY/T 0863Ў¶ТҪУГДЪҝъҫөДЪҝъҫө№ҰДЬ№©ёшЧ°ЦГ№цС№КҪіеПҙОьТэЖчЎ·өИұкЧјЦЖ¶ЁЎЈҫӯДЪҝъҫөНЁөАҪшИлИЛМеөД»№УҰҝјВЗGB 9706.19ұкЧјөДПа№ШТӘЗуЎЈ
ЎЎЎЎ2. ЙъОпС§МШРФСРҫҝ
ЎЎЎЎУҰ¶ФІъЖ·Ҫб№№ЧйіЙЦРУл»јХЯҪУҙҘІҝ·ЦөДЙъОпПаИЭРФҪшРРЖАјЫЎЈЙъОпПаИЭРФЖАјЫҝЙёщҫЭGB/T 16886.1-2011Ў¶ТҪБЖЖчРөЙъОпС§ЖАјЫөЪ1Іҝ·ЦЈә·зПХ№ЬАн№эіМЦРөДЖАјЫУлКФСйЎ·әНЎ¶№ШУЪУЎ·ўТҪБЖЖчРөЙъОпС§ЖАјЫәНЙуЖАЦёДПөДНЁЦӘЎ·(№ъКіТ©јаРөЎІ2007Ўі345әЕ)өДТӘЗуҪшРРЎЈЙъОпПаИЭРФЖАјЫСРҫҝЧКБПУҰөұ°ьАЁЈәЙъОпПаИЭРФЖАјЫөДТАҫЭәН·Ҫ·Ё;ІъЖ·ЛщУГІДБПөДГиКцј°УлИЛМеҪУҙҘөДРФЦК;КөК©»т»нГвЙъОпС§КФСйөДАнУЙәНВЫЦӨ;¶ФУЪПЦУРКэҫЭ»тКФСйҪб№ыөДЖАјЫЎЈ
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІбҙъАнЙкЗлИЛМṩЙъОпПаИЭРФЖАјЫСРҫҝЧКБПКұЗл№ШЧўТФПВ·ҪГжЈә
ЎЎЎЎ(1)ЙъОпПаИЭРФЖАјЫУҰ¶ФіЙЖ·¶шІ»КЗФӯІДБПҪшРРЖАјЫЈ¬Іҝ·ЦІДБПЙъІъјУ№Ө№эіМҝЙДЬ»бёДұдІДБПөДЙъОпПаИЭРФҪб№ыЈ¬АэИзМнјУБЛјУ№ӨЦъјБЎў»тХЯјУ№Ө№эіМ(АэИзёЯОВ)ёДұдБЛФӯІДБПөДРФЦКЎЈ
ЎЎЎЎ(2)ЙъОпС§КФСй°ҙХХЎ¶№ШУЪКөК©Ў¶ТҪБЖЖчРөЧўІбУлұё°ё№ЬАн°м·ЁЎ·Ў¶МеНвХп¶ПКФјБЧўІбУлұё°ё№ЬАн°м·ЁЎ·УР№ШКВПоөДНЁёжЎ·(№ъјТТ©Ж·ја¶Ҫ№ЬАнҫЦ2021ДкөЪ76әЕ)өДТӘЗуМбҪ»ЧКБПЎЈ
ЎЎЎЎ(3)ЙъОпС§КФСйұЁёжУҰМеПЦІъЖ·ГыіЖәНРНәЕЈ¬УлЙкұЁІъЖ·Па¶ФУҰЎЈИзМṩЖдЛыІъЖ·өДұЁёжЈ¬УҰ¶ФКФСйІъЖ·УлЙкұЁІъЖ·өДІоТмРФҪшРРЖАјЫЈ¬УҰЦӨГчФӯІДБПј°АҙФҙЎўЙъІъ№ӨТХөИУ°ПмЙъОпПаИЭРФЦШРВЖАјЫөДІоТмҫщІ»ҙжФЪЎЈ
ЎЎЎЎ(4)ҪрКфІДБПИфІЙУГБЛ№ъјТ»тРРТөұкЧјЦРУҰУГ·¶О§ККәПөДТҪУГҪрКфІДБПЈ¬ҝЙІ»ҪшРРЙъОпС§КФСйЎЈө«РијмІвИП¶ЁІДЦКЎЈ
ЎЎЎЎ3.Пы¶ҫГрҫъ№ӨТХСРҫҝ
ЎЎЎЎКЦұъј°№ӨҫЯН·К№УГЗ°УҰГрҫъЎЈ
ЎЎЎЎУЙК№УГХЯ¶ФКЦұъЎў№ӨҫЯН·өИҪшРРЗеПҙЎўПы¶ҫЎўГрҫъөДЈ¬УҰөұГчИ·НЖјцөД№ӨТХ(·Ҫ·ЁЎўІОКэ)ј°И·¶ЁөДТАҫЭЈ¬ІўМṩЗеПҙЎўПы¶ҫЎўГрҫъөДСРҫҝЧКБПЎЈ¶ФҝЙДНКЬБҪҙО»т¶аҙОГрҫъөДІъЖ·Ј¬ёЕКцЛщМṩІъЖ·¶ФГрҫъ·Ҫ·ЁДНКЬРФөДСРҫҝЧКБПЗйҝцЎЈ
ЎЎЎЎУЙЙъІъЖуТөГрҫъәуҪ»ё¶өДТ»ҙОРФК№УГөД№ӨҫЯН·ЎўКЦұъЈ¬УҰГчИ·Грҫъ№ӨТХ(·Ҫ·ЁәНІОКэ)әНОЮҫъұЈЦӨЛ®ЖҪ(SAL)Ј¬ІўМṩГрҫъИ·ИПұЁёжЎЈ¶ФУЪІЙУГ·шХХГрҫъөДЈ¬УҰөұМṩЧоҙуҝЙҪУКЬјББҝЎўГрҫъјББҝТФј°јББҝ·ЦІјөДСРҫҝЎЈ¶ФУЪІЙУГ»·СхТТНй(EO)өИТЧІъЙъІРБфөДГрҫъ·ҪКҪЈ¬УҰөұМṩІРБфОпөДЧоҙуІРБфЛ®ЖҪј°ЖдСРҫҝЧКБПЎЈ
ЎЎЎЎ4.ІъЖ·УРР§ЖЪЎў°ьЧ°СРҫҝ
ЎЎЎЎІъЖ·УРР§ЖЪТ»°гИЎҫцУЪК№УГ№эіМЦРІҝјюЎўФӘЖчјюөДЛрәДЎўАП»ҜөИЈ¬ТҪБЖЖчРөЧўІбЦӨ°мАнЙкЗлИЛҝЙ°ҙХХЙщіЖөДУРР§ЖЪ¶ФЙиұёҪшРРАП»Ҝ/ЖЈАНКФСйЈ¬ТІҝЙ¶ФУ°ПмЙиұёУРР§ЖЪөД№ШјьІҝјюҪшРРАП»Ҝ/ЖЈАНКФСйЈ¬АэИзЙиұёЦРІ»ҝЙёь»»(»тёь»»іЙұҫёЯ)өДІҝјюЈ¬МṩПаУҰөДСРҫҝЧКБПЎЈДЪҝъҫөКЦКх¶ҜБҰЙиұёЦРЦч»ъЎўКЦұъЎўҪЕМӨҝӘ№ШөИУРФҙІҝјюИфНЁ№эјУЛЩАП»ҜКФСй(ІОҝјGB /T 34986-2017)ҪшРРСйЦӨөДЈ¬КФСйУҰЦБЙЩҝјВЗОВ¶ИәНКӘ¶ИБҪёцУҰБҰЛ®ЖҪЈ¬»№УҰҝјВЗөзЧУФӘЖчјюТФј°ІДБПөДАП»ҜЎўІЩЧчК№УГДҘЛр(№ӨҫЯН·ЎўКЦұъБ¬ҪУІҝөИ)ЎўПы¶ҫГрҫъ·ҪКҪөИ¶ФК№УГДкПЮөДУ°ПмЎЈСРҫҝЧКБПУҰДЬЦӨГчІъЖ·°ҙХХЛщЙщіЖөДУРР§ЖЪЈ¬ҫӯ№эАП»Ҝ/ЖЈАНКФСйәуЈ¬ІъЖ·РФДЬәН°ІИ«ИФ·ыәПФӨЖЪөДТӘЗуЎЈ
ЎЎЎЎУРР§ЖЪТІҝЙ»щУЪТСУРКэҫЭҪшРРәПАнөДНЖ¶ПЎў·ЦОцЎўјЖЛгөГіцЈ¬ө«УҰМṩПкПёөДЛөГчј°Ц§іЦРФЧКБПЎЈҫЯМеҝЙІОҝјЎ¶УРФҙТҪБЖЖчРөК№УГЖЪПЮЧўІбјјКхЙуІйЦёөјФӯФтЎ·өДТӘЗуЎЈ
ЎЎЎЎОЮҫъ°ьЧ°өД№ӨҫЯН·ЎўКЦұъЈ¬Жд»хјЬУРР§ЖЪәН°ьЧ°НЁ№эКФСйҪшРРСйЦӨөДЈ¬КФСй·Ҫ·ЁҝЙІОҝјYY/T 0681ПөБРұкЧјЎЈІ»Н¬Грҫъ·ҪКҪЎўІ»Н¬°ьЧ°ІДБПөДІъЖ·Ј¬УҰ·ЦұрҪшРРУРР§ЖЪСйЦӨәН°ьЧ°СРҫҝЎЈ
ЎЎЎЎҝЙЦШёҙК№УГөД№ӨҫЯН·ЎўКЦұъөДК№УГКЩГьИфУлЗеПҙЎўГрҫъҙОКэПа№ШЈ¬»№УҰМбҪ»ПаУҰөДОИ¶ЁРФј°ДНКЬРФСйЦӨ·ЦОцұЁёжЈ¬ЦБЙЩУҰөұЦӨГчЦШёҙК№УГ№эіМЦРЈ¬ІЙУГПаУҰөДЗеПҙЎўГрҫъ№ӨТХҙҰАнәуЈ¬ЙкұЁІъЖ·ФЪУРР§ЖЪДЪКЗ°ІИ«УРР§өДЎЈІўёщҫЭ·ЦОцұЁёжЈ¬И·¶ЁҝЙЦШёҙК№УГҙОКэЎЈ
ЎЎЎЎ°ьЧ°СРҫҝЧКБПУҰ¶ФІъЖ·ј°°ьЧ°ҪшРРДЈДвКФСйЈ¬ДЈДвФЪЦьҙжәНФЛКд№эіМЦРЈ¬УцөҪёҙФУЗйҝцКұЈ¬АэИз»·ҫі(ОВКӘ¶ИЎўЖшС№өИ)ұд»ҜЎўөшВдЎўХс¶ҜөИЈ¬ІъЖ·І»»б·ўЙъРФДЬЎў№ҰДЬЎў°ІИ«РФёДұдЈ¬°ьЧ°ҫЯУРұЈ»ӨІъЖ·ДЬБҰЎЈҫӯ№эДЈДвКФСйә󣬹۲м°ьЧ°Нв№ЫКЗ·сУРІ»ҝЙҪУКЬөДТміЈЗйҝц;¶ФІъЖ·ҪшРРРФДЬІвКФЈ¬ЦӨГчФЛКдәН»·ҫіІвКФәуІъЖ·ДЬ№»ұЈіЦЖдНкХыРФЎў№ҰДЬРФәН°ІИ«РФЎЈ
ЎЎЎЎ5.ИнјюСРҫҝ
ЎЎЎЎИфІъЖ·ә¬УРИнјюЈ¬ТҪБЖЖчРөІъЖ·ЧўІбЦӨ°мАнЙкЗлИЛУҰ°ҙХХЎ¶ТҪБЖЖчРөИнјюЧўІбјјКхЙуІйЦёөјФӯФтЎ·өДТӘЗуМṩПаУҰөДИнјюСРҫҝЧКБПЎЈН¬КұЈ¬УҰөұіцҫЯ№ШУЪИнјю°жұҫГьГы№жФтөДЛөГчЈ¬ГчИ·Инјю°жұҫөДИ«ІҝЧЦ¶Ој°ЧЦ¶Оә¬ТеЈ¬И·¶ЁИнјюөДНкХы°жұҫәН·ўІј°жұҫЎЈ
ЎЎЎЎёГАаІъЖ·НЁіЈІ»ҫЯУРІЙУГҙжҙўГҪҪй»тә¬УРНшВзБ¬ҪУ№ҰДЬТФҪшРРөзЧУКэҫЭҪ»»»»тФ¶іМҝШЦЖ№ҰДЬЈ¬ИфУРЈ¬ФтЙкЗлИЛУҰ°ҙХХЎ¶ТҪБЖЖчРөНшВз°ІИ«ЧўІбјјКхЙуІйЦёөјФӯФтЎ·өДТӘЗуМṩПаУҰөДСРҫҝЧКБПІўФЪјјКхТӘЗуЦРФцјУКэҫЭҪУҝЪЎўУГ»§·ГОКҝШЦЖөИРЕПўЎЈ
ЎЎЎЎ(ЛД)·зПХ·ЦОц
ЎЎЎЎ°ҙХХYY/T 0316Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·өДТӘЗ󣬶ԲъЖ·ЙъГьЦЬЖЪИ«№эіМКөК©·зПХ№ЬАнЎЈФЪІъЖ·ЧјұёЧўІбЙПКРЗ°Ј¬УҰ¶Ф·зПХ№ЬАн№эіМҪшРРЖАЙуЎЈЖАЙуУҰЦБЙЩИ·ұЈЈә·зПХ№ЬАнјЖ»®ТСұ»ККөұөШКөК©;ЧЫәПКЈУа·зПХКЗҝЙҪУКЬөД;ТСҪЁБўІъЖ·ЙПКРәуөДЧ·ЛЭУлБЩҙІУҰУГРЕПўКХјҜЦЖ¶ИЎЈ
ЎЎЎЎІъЖ·өД·зПХ№ЬАнұЁёжУҰ·ыәПYY/T 0316өДУР№ШТӘЗуЈ¬ЕР¶ПУлІъЖ·УР№ШөДОЈПХФҙЈ¬№АјЖәНЖАјЫПа№Ш·зПХЈ¬ҝШЦЖХвР©·зПХІўјаКУҝШЦЖөДУРР§РФЎЈПа№ШөДОЈәҰ°ьАЁДЬБҝОЈәҰЎўЙъОпС§ОЈәҰЎў»·ҫіОЈәҰЎўУлІЩЧчК№УГУР№ШөДОЈәҰЎўИнјюөДОЈәҰЎўИЛ»ъ№ӨіМС§ОЈәҰЎў№ҰДЬК§Р§ЎўО¬»ӨәНАП»ҜөјЦВөДОЈәҰөИ·ҪГжЎЈ
ЎЎЎЎ¶ФУЪіеПҙЎў№аЧўЎўОьТэ№ҰДЬЈ¬·зПХҝШЦЖЧКБПЦРҪЁТйҝјВЗЛ®өзҪвЦКОЙВТЎўЖшЛЁЎўЗ»МеЛъПЭөИҙшАҙөД°ІИ«РФ·зПХЎЈАэИзЈ¬ИфЕЩПчЙиұёөДОьТэБчЛЩ№эҙуЈ¬УлЕдәПК№УГөДЖдЛы№аБчЙиұёөДБчБҝ¶МКұІ»ЖҘЕдЈ¬»бФміЙЗ»МеЛъПЭЈ¬КЦКхКУТ°ұдІоҪш¶шөјЦВКЦКхОЮ·ЁјМРшҪшРРЈ¬ИфҙЛКұЕЩПчН·ИФФЪРэЧӘ№ӨЧчФт»бҙжФЪҪПёЯөД°ІИ«·зПХЎЈ
ЎЎЎЎ(Ое)ІъЖ·јјКхТӘЗу
ЎЎЎЎІъЖ·јјКхТӘЗуУҰ°ҙХХЎ¶ТҪБЖЖчРөІъЖ·јјКхТӘЗуұаРҙЦёөјФӯФтЎ·өДТӘЗуҪшРРұаРҙЎЈјјКхТӘЗуЦРЦчТӘ°ьАЁІъЖ·ЧФЙнРФДЬөДІОКэЎўИнјю·ўІј°жұҫј°ГьГы№жФтТФј°НшВз°ІИ«(ИфУР)ТӘЗуөИДЪИЭЎЈә¬УР¶аёц№жёсРНәЕөДЈ¬УҰГчИ·РНәЕЦ®јдөДІоТмЎЈ
ЎЎЎЎТҪБЖЖчРөЧўІбЦӨҙъАнЙкЗлИЛУҰІОҝјПаУҰөД№ъјТұкЧјЎўРРТөұкЧјЈ¬ІўҪбәПБЩҙІРиЗуЎўЧФЙнІъЖ·өДјјКхМШөг¶ФёчПоЦёұкөДҫЯМеІОКэЧчіц№ж¶ЁЎЈЕЩПчН·ҝЙІОҝјРРТөұкЧјYY/T0955Ў¶ТҪУГДЪҝъҫөДЪҝъҫөКЦКхЙиұёЕЩПчЖчЎ·ЦЖ¶ЁРФДЬЦёұкөДТӘЗуЈ¬ЖдЛы№ӨҫЯН·ТІҝЙІОҝјёГұкЧјЎЈ
ЎЎЎЎУЙУЪДҝЗ°YY/T0955ұкЧјЦРөДКФСй·Ҫ·ЁҪцККУГУЪЕЩПчН·Ј¬ЖдЛы№ӨҫЯН·(ИзДҘН·ЎўЧкН·өИ)ёЯЧӘЛЩКұёәФШМШРФөДІвБҝІўІ»ККУГЈ¬ЙъІъЖуТөҝЙёщҫЭІъЖ·ЙијЖЧФРРЦЖ¶ЁёЯЧӘЛЩКұөДёәФШМШРФөДКФСй·Ҫ·ЁЈ¬ө«РиМṩКФСй·Ҫ·ЁәПАнРФөДЦ§іЦРФЧКБПЎЈ
ЎЎЎЎИфІъЖ·ҫЯУРіеПҙөИМШКв№ҰДЬЈ¬ЙкЗлИЛ»№УҰёщҫЭІъЖ·ЧФЙнМШРФ(ИзҪөОВЎўЗеҙҙ)ФЪјјКхТӘЗуЦР№ж¶ЁМШКв№ҰДЬПаУҰөД¶ЁРФ»т¶ЁБҝөДТӘЗуЎЈ
ЎЎЎЎУЙУЪёГАаІъЖ·өД№ӨҫЯН·ҙу¶аОӘҪрКфІДБПЈ¬јјКхТӘЗуЦРУҰГчИ·Ул»јХЯЦұҪУ»тјдҪУҪУҙҘөДҪрКфІДБПЈ¬ІўұкГчЛщСЎҪрКфІДБПөДЕЖәЕәН/»тҙъәЕЎЈ
ЎЎЎЎ(Бщ)јмСйұЁёж
ЎЎЎЎ1.¶ФУЪН¬Т»ёцТҪБЖЖчРөЧўІбөҘФӘҫЯУР¶аёц№жёсРНәЕөДЈ¬ФӯФтЙПСЎИЎҪб№№ЧоёҙФУЎў№ҰДЬЧо¶аЎўЦёұкЧоёЯөДЧчОӘөдРНРНәЕҪшРРРФДЬәНөзЖш°ІИ«јмІвЈ¬ІўМṩПкПёөДөдРНРФ·ЦОцЎЈ
ЎЎЎЎ2.өзҙЕјжИЭұЁёжЦРІъЖ·»щұҫРЕПў(өзС№ЎўЖөВКөИ)Ўў·ЦЧй·ЦАаУҰУлІъЖ·јјКхТӘЗуЎўЛөГчКйөИПа№ШДЪИЭТ»ЦВЎЈ№ӨЧчДЈКҪУҰСЎФсЧоІ»АыЗйҝцЈ¬АэИзЧоҙуЧӘЛЩөИЎЈДЪҝъҫөКЦКх¶ҜБҰЙиұё»щұҫРФДЬНЁіЈҝЙОӘЈәХэіЈЧӘЛЩЎўПФКҫЎўОьТэ(Ифә¬)ЎЈ
ЎЎЎЎ(ЖЯ)ЖдЛыЧКБП
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІбЙкЗлДЪҝъҫөКЦКх¶ҜБҰЙиұёТСБРИлЎ¶ГвУЪБЩҙІЖАјЫТҪБЖЖчРөДҝВј(2021Дк)Ў·(ТФПВјтіЖЎ¶ДҝВјЎ·Ј¬№ъјТТ©Ж·ја¶Ҫ№ЬАнҫЦНЁёж2021ДкөЪ71әЕ)өЪ24ПоЈ¬ҝЙГвУЪМбҪ»БЩҙІЖАјЫЧКБПЎЈЙкЗлИЛУҰөұ°ҙХХЎ¶БРИлГвУЪҪшРРБЩҙІЖАјЫТҪБЖЖчРөДҝВјІъЖ·¶ФұИЛөГчјјКхЦёөјФӯФтЎ·Ј¬ҙУ»щұҫФӯАнЎўҪб№№ЧйіЙЎўРФДЬЎў°ІИ«РФЎўККУГ·¶О§өИ·ҪГжЈ¬ЦӨГчІъЖ·өД°ІИ«УРР§РФЎЈ
ЎЎЎЎИфОЮ·ЁЦӨГчЙкұЁІъЖ·УлЎ¶ДҝВјЎ·ІъЖ·ҫЯУРөИН¬РФЈ¬ФтУҰМбҪ»БЩҙІЖАјЫЧКБПЎЈ
ЎЎЎЎ¶ФУЪТ»ҙОРФК№УГөД№ӨҫЯН·Ј¬»№УҰөұМṩЦӨГчЖдОЮ·ЁЦШёҙК№УГөДЦ§іЦРФЧКБПЎЈ
ЎЎЎЎ(°Л)ЛөГчКйәНұкЗ©
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІб°мАнЙкЗлЛөГчКйәНұкЗ©УҰ·ыәПЎ¶ТҪБЖЖчРөЛөГчКйәНұкЗ©№ЬАн№ж¶ЁЎ·(Фӯ№ъјТКіЖ·Т©Ж·ја¶Ҫ№ЬАнЧЬҫЦ6әЕБо)ЎўYY/T 0955ј°ЖдЛыККУГұкЧјЦРУР№ШЛөГчКйәНұкЗ©өДТӘЗуЈ¬УҰ°ьАЁУРР§ЖЪЎўЛөГчКйРЮ¶©»тұаЦЖИХЖЪөИПёҪЪЎЈИфІъЖ·ә¬УРУРПЮК№УГҙОКэөД№ӨҫЯН·»тКЦұъЈ¬»№УҰФЪЛөГчКйЦРГчИ·ҝЙЦШёҙК№УГІҝјюөДК№УГҙОКэЈ¬ІўМбКҫІ»ДЬјМРшК№УГөДЗйРОЎЈТ»ҙОРФК№УГөД»№УҰГчИ·»хјЬУРР§ЖЪЎЈ
ҪшҝЪЎў№ъДЪТҪБЖЖчРөІъЖ·ЧўІбј°ЙъІъРнҝЙ°мАн(°ьАЁЈәТҪБЖЖчРөІъЖ·ЧўІбјјКхОДјюұаРҙЎўТҪБЖЖчРөЦКБҝ№ЬАнМеПөҪЁБўЎўПЦіЎМеҝјәЛІйЦёөјЎўЙијЖҝӘ·ўОДјюұаРҙЦёөјЎўЕъјЗВјұаРҙЦёөјЎўПЦіЎЕаСөЎўЙъІъРнҝЙЙкұЁЧКБПұаРҙј°Па№ШКВПоНшЙППөНіЙкұЁөИ)ЎўТ»АаТҪБЖЖчРөІъЖ·ұё°ёЎўЙъІъұё°ёЎў№гёжЕъОДЎўіцҝЪПъКЫЦӨГч°мАнЈ¬Ҫаҫ»КТҪЁЙи№ж»®өИКВПоРиТӘРӯЦъёЁөј°мАнЗлДъХТЙоЫЪәиФ¶ТҪБЖЖчРөЧЙСҜУРПЮ№«ЛҫОӘДъМṩһХҫКҪЧЁТө·юОсЈ¬»¶УӯЧЙСҜЈЎ
ЎЎЎЎ
|

