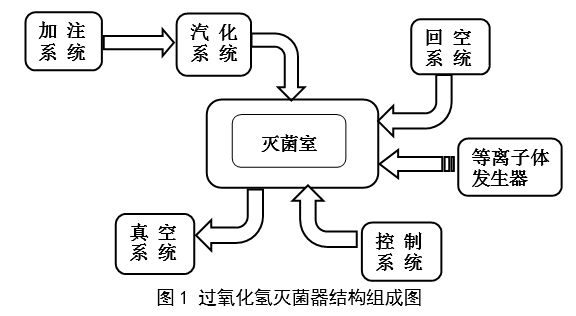
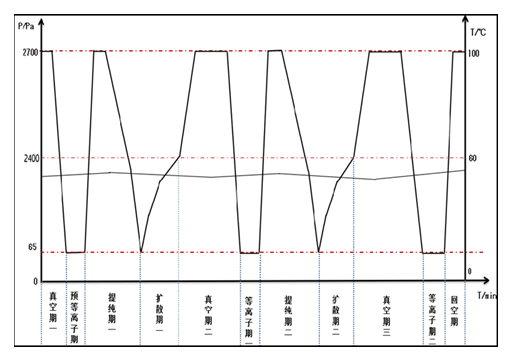
|
电磁能 |
保护接地阻抗、可触及部件允许限值 |
电源故障时设备上会有电压差,人员接触会形成电流回路 |
电损伤,严重时死亡 |
|
设备漏电使设备外表面带电 |
操作人员接触到带电部分 |
电损伤,严重时死亡 |
|
机器外壳的防护罩封闭不良 |
可接触到带电部分 |
电损伤,严重时死亡 |
|
环境设施电源干扰、电池容量不足、带静电的操作者触摸操作按键、屏幕等,造成显示电子器件损坏、错误显示或程序异常 |
设备运行异常或不能工作 |
灭菌失败 |
|
等离子电源输出为高频高压电源,常规仪器无法检测 |
若防护不当,人员可能会接触到带电部分 |
电损伤,严重时死亡 |
|
热能 |
测温、控时部件损坏,控制失灵 |
设备温度超出限定值、时间不到设定值 |
物品损坏,人员受伤 |
|
操作者接触设备温度部件 |
接触高温器件表面 |
人员受伤 |
|
机械能 |
门密封失效,密封不严,门锁机构失效 |
有毒气体泄漏或门不能打开 |
环境污染,灭菌失败 |
|
外壳粗糙、有毛刺 |
操作人员划伤,损伤灭菌器械 |
人员受伤,物品损坏 |
|
运输过程中外壳受外力碰撞,造成腔体破裂,门挤压变形 |
设备或部件损坏 |
无法使用 |
|
设备压力未在规定值范围,压力监测装置失效 |
设备不能正常工作 |
物品损坏,灭菌失败 |
|
设备消音系统或运动部件损坏 |
噪声超出限定值 |
人员不适 |
|
化学 |
容器壳体泄漏、管路泄漏、过滤器失效、操作不当等引起过氧化氢泄露 |
过氧化氢气体泄漏到环境中 |
皮肤、眼睛过敏,灭菌物品损坏,灭菌失败 |
|
排放过量的过氧化氢气体 |
对人员造成伤害 |
人员灼烧、过敏 |
|
过氧化氢注入量过多 |
过氧化氢残留超标 |
人员灼烧、过敏,物品损坏 |
|
过氧化氢注入量过少 |
灭菌不彻底 |
灭菌失败 |
|
灭菌负载表面冷凝 |
冷凝表面会含有过量过氧化氢,人员接触,造成伤害 |
人员灼烧、过敏,物品损坏 |
|
操作 |
对不适用的物品进行灭菌或选用了不匹配的灭菌周期 |
物品不能灭菌,受到腐蚀 |
物品损坏,灭菌失败 |
|
使用人员操作不熟练、使用不当,负载未经彻底干燥或者装载量过大 |
抽真空超时,灭菌程序终止;过氧乙酸冷凝;负载接触过氧化氢减少 |
灭菌失败 |
|
操作过程过于复杂,使用操作时失误 |
不能正常使用 |
灭菌失败 |
|
不完整的说明书 |
说明书中有关维护、保养等内容不明确。如:清洗、预防性检查、保养周期等 |
不能及时维护,更换易耗部件 |
产品故障,性能下降,寿命降低 |
|
说明书中缺少必要的警告说明和详细的使用方法 |
错误操作 |
设备无法正常使用,人员受伤 |
|
未明确可更换部件的更换周期,如真空油泵、油雾过滤器、过氧化氢过滤器、空气过滤器 |
产品或部件超期使用 |
产品出现故障,性能下降,残留超标 |
|
|
外部和内部标记不全面、标记不正确或不能够清楚易认,标记位置不恰当,以及标记不能够永久贴牢 |
错误使用;
产品辨别错误。 |
设备无法正常使用 |