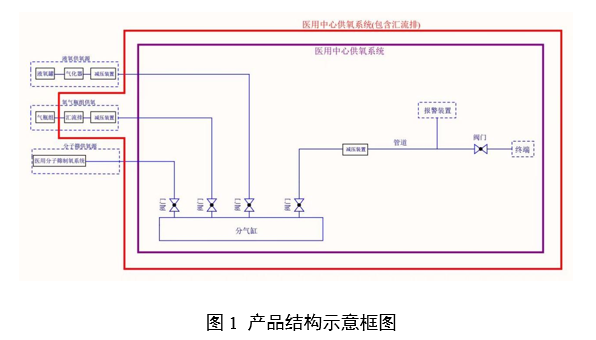
|
ЕчДХФмСП |
дкЧПЕчДХЗјЩфдДБпЪЙгУЕчЖЏЩшБИЃЌПЩФмЛсГіЯжЕчДХИЩШХГЬађдЫааЃЌЕМжТГЬађДэЮѓ |
|
ЕчбЙВЈЖЏЃЌПЩФмЕМжТе§дкЙЄзїжаЕФЩшБИГіЯжбѕЦјбЙСІСїСПБфЛЏ |
|
ЛњаЕФм |
ГЄЪБМфМАЙ§ИпЕФЙЉбѕбЙСІЃЌПЩФмЕМжТжеЖЫМАЦфИНМўЕФЫ№ЛЕ |
|
Й§ЕЭЕФЙЉбѕбЙСІЃЌПЩФмЛсГіЯжЮоЗЈЭЈЦј/е§ГЃЙЉЦјЃЌЕМжТЮоЗЈГіЦјЛђДяЕНбЙСІвЊЧѓЃЌВЛФмИјЛМепе§ГЃЙЉЦј |
|
ЛЏбЇЮЃКІ |
ЙмЕРЭбжЌЧхЯДВЛЭъШЋЃЌЙмЕРжаЕФгЭжЌЕШЛьЕНбѕЦјжаЃЌЬМЧтЛЏКЯЮяКЌСПГЌБъЃЌЙЉЛМепЪЙгУЕФбѕЦјжЪСПВЛКЯИёЃЌЧвДцдкИаШОЗчЯе |
|
ЪЙгУВЛКЯИёЕФвНгУбѕЛђИЛбѕПеЦјЃЈ93%бѕЃЉЃЌЪЙгУЮДгавЉЦЗзЂВсжЄЕФвНгУбѕЃЌЛђЪЙгУЮДОзЂВсЕФвНгУЗжзгЩИжЦбѕЯЕЭГзїЮЊбѕдДЃЌдьГЩбѕЦјжЪСПВЛДяБъ |
|
ВФСЯЕФбЁдёЮДПМТЧгыбѕЦјЕФМцШнадЃЛН№ЪєВФСЯЮДПМТЧдкжиДѓв§ШМЛЗОГФмЗёБЛв§ШМЃЛШМЩеЕФгаЖОЮяжЪЛђЗЧН№ЪєВФСЯЃЈАќРЈШѓЛЌМСЃЌШчЪЙгУЃЉЕФЗжНтКЭЧБдкЮлШОЮяОЙмТЗНјШыШЫЬхЕФЗчЯе |
|
ЙІФмЕФЩЅЪЇЛђБфЛЕ |
ЙмЕРВПЗжЛђЭъШЋЖТШћЃЌЛМепЛђЩшБИЕФЙЉбѕЫ№ЪЇЛђМѕЩй |
|
ЪЙгУВЛКЯИёЕФбЙСІШнЦїЃЌШчЪЙгУЮДгаЬижжЩшБИаэПЩжЄЕФвКбѕЙоЃЈАќРЈвНгУбѕКИНгОјШШЦјЦПЃЉЁЂбѕЦјЦПЃЌдьГЩаЙТЉЁЂЮоЗЈе§ГЃЙЉбѕ |
|
дЫаажаЕФбѕдДЙЉЦјМѕЩйЃЌШчЙћжївЊЦјдДКЭДЮвЊЦјдДЗЂЩњЙЪеЯЃЌБИгУЛђгІМБЦјдДЮЊЯЕЭГЙЉЦјЁЃШчЙћЫљгаЯЕЭГЖМЗЂЩњЙЪеЯЃЌжеЖЫЩшБИЕФбѕЦјЙЉЦјЪЇаЇ |
|
ШчЙћвЛБИвЛгУЯрЛЅЧаЛЛТпМВЛФме§ГЃЪЕЪЉЃЌгІИУгаЯргІЬсЪОвдБуЪжЖЏЧаЛЛ |
|
ЙмЕРЗЂЩњджФбадЙЪеЯЃЌдьГЩжеЖЫЙЉбѕВЛзу |
|
БЈОЏЙЪеЯ/БЈОЏЯЕЭГЖЯЕчЃЌЧБдкЙЉбѕЙЪеЯЪБЮДБЈОЏ |
|
ЕчЦјВПМўЕФдЫааЙЪеЯПЩФмЛсЕМжТЙЉЕчжаЖЯ |
|
ЙиМќВПМўГіЯжЙЪеЯЃЌЦјдДДцдкЧБдкЫ№ЪЇ |
|
ЙмЭјаЙТЉЃЌдьГЩЧБдкЕФЛ№джЗчЯеЁЂДцдкжЯЯЂЕФЧБдкЗчЯеЁЂжеЖЫЙЉбѕВЛзуЛђМѕЩй |
|
ЪЙгУСНжжВЛЭЌжЪСПЕФбѕдДЃЈ93бѕЦјгыДПбѕЃЉЃЌЪгЛМепЕФЧщПіЖјЖЈЃЌбѕЦјХЈЖШПЩФмЛсЖдЛМепЕФжЮСЦВњЩњВЛРћгАЯь |
|
зЙТф |
вђдкдЫЪфЙ§ГЬжаВЛаЁаФЛђдкаЖЛѕЪБВЛаЁаФНЋЯЕЭГзщМўЫЄЛЕЛђзВЛЕЃЌПЩФмГіЯжвђБфаЮЖјВЛФме§ГЃЪЙгУЃЌЕМжТВњЩњТЉЦјЛђВЛФмГіЦјЯжЯѓЃЌВЛФмИјЛМепе§ГЃЙЉЦј |
ВЛе§ШЗЕФ
ВтСП |
бЙСІБэЮДОаЃзМЛђепЫ№ЛЕЃЌГіЯжбЙСІЪфГіЦЋВюЃЌЕМжТЪфГіЙ§ИпЛђЙ§ЕЭЕФбѕЦјбЙСІ |
|
ВйзїДэЮѓ |
ВПМўКЭЙмЕРЯЕЭГЕФЩшМЦЛђАВзАВЛЕБЃЌЙмЕРЯЕЭГЙЉбѕВЛзу |
|
ЪЙгУДэЮѓЃКЮДОХрбЕЕФШЫдБЪЙгУЙЉбѕЩшБИЃЌПЩФмГіЯжДэЮѓЕиЩшЖЈЙЉбѕбЙСІжЕКЭДэЮѓЕиВйзїЙЉбѕЩшБИЃЌЕМжТЪфГіЙ§ИпЛђЙ§ЕЭЕФбѕЦјбЙСІ |
|
ЩшБИВ№аЖКѓЮДФме§ШЗзщзАЃЌШчПеЦјЙ§ТЫЦїЁЂЙЉбѕЙмЕРЕФДэЮѓСЌНгЃЌМАЮДдкЪЙгУЧАНјааЙІФмВтЪдЃЌГіЯжЮоЗЈе§ГЃЙЉбѕЃЌЕМжТвКЬх/ЙЬЬхвчГіЃЌНјШыжаМфЙмТЗ |
|
дкзЂВсЩъЧыШЫЙцЖЈЕФЪЙгУЛЗОГЬѕМўЭтЪЙгУВњЦЗЃЌГіЯжЩшБИадФмЯТНЕЁЂЪЇаЇЃЌЕМжТЛМепЕУВЛЕНгааЇжЮСЦ |
|
ЪЙгУВЛе§ШЗЕФИНМўЃЌГіЯжЙЉбѕЙІФмЪЇаЇЃЌЕМжТЛМепЕУВЛЕНжЮСЦ |
|
ЮДЪЕЪЉЙцЖЈЕФБЃбјЃЈШчЖдгагЭШѓЛЌБУМгзЂШѓЛЌгЭЃЉЃЌГіЯжЩшБИадФмЯТНЕЁЂЪЇаЇЃЌЕМжТВњЦЗадФмСгЛЏЃЌбЯжиЪБЩеЛй |
|
ЮДзіКУбеЩЋМАНгПкЧјБ№ЕФЙЉбѕЖЫПкШнвзВхДэЃЌПЩФмЕМжТдкМБОШЕФНєМБзДПіЯТЃЌгУбѕМАЙЉбѕЕФЪЙгУГіЯжВхДэ |
|
вђГЄЦкЪЙгУЃЌВПЗжИНМўГіЯжЫЩЖЏЛђзшШћЯжЯѓЃЌПЩФмГіЯжТЉЦјЛђВЛЭЈЦјЃЌЕМжТЛМепвЊЪЙгУЪБЃЌЮоЦјПЩгУ |
|
ЮДВЩгУгХжЪЕФИНМўЃЌдКЗНдкЪЙгУЪБЗНЗЈВЛЕУЕБЃЌВЛПЩБЃжЄЦфЪЙгУЪйУќЃЌПЩФмГіЯжТЉЦјЛђВЛЭЈЦјЃЌЕМжТЪЙгУЪйУќЬсЧАжежЙ |
ВЛЭъећЕФ
ЫЕУїЪщ |
ЮДЖдДэЮѓВйзїНјааЫЕУїЃЌПЩФмГіЯжВйзїДэЮѓЃЌЕМжТЛМепЕУВЛЕНжЮСЦЃЌзщжЏЃЌеГФЄЫ№ЩЫЁЃ |
|
ВЛе§ШЗЕФЯћЖОЗНЗЈЃЌПЩФмГіЯжЪЙгУгаИЏЪДадЕФЧхНрМСЁЂЯћЖОМСЃЌЕМжТВњЦЗВПМўИЏЪДЃЌЗРЛЄадФмНЕЕЭ |
|
ВЛе§ШЗЕФВњЦЗжќДцЬѕМўЃЌПЩФмГіЯжЦїМўРЯЛЏЃЌВПМўЪйУќНЕЕЭЃЌЕМжТВњЦЗадФмНЕЕЭЃЌЪйУќЫѕЖЬ |
|
ЮДЙцЖЈаЃбщжмЦкЃЌПЩФмГіЯжбЙСІБэЦЋВюЃЌЕМжТВЛе§ШЗЕФВтСП |