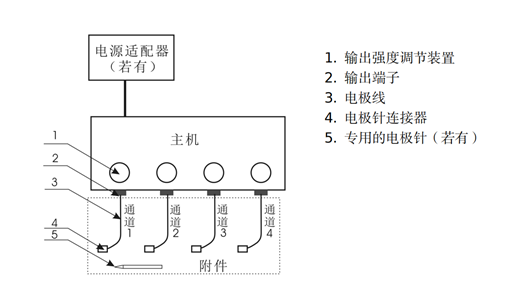
|
µзґЕДЬБїЈЁПЯµзС№Ј© |
µзј«ПЯОЮТвµШІеИлБЛНшµзФґІеЧщ |
НшµзС№іцПЦФЪХлЙП |
СПЦШЙХЙЛЎўРДФаІь¶ЇЎўЛАНц |
|
µзґЕДЬБїЈЁESDЈ© |
ІъЖ·µДРЕєЕ·ўЙъєНЗї¶ИїШЦЖіцПЦ№КХП |
ІъЙъБЛёЯДЬБїµДВціеКдіц |
ФміЙµз»чЙЛє¦ |
|
»ъРµДЬБї |
µзј«ПЯ№эґЦ№эУІ |
Ѕ«ХлАЖ«ЎўА¶ПЈ¬ФміЙУлБЩЅьµДХл¶МВ·Ј¬ФміЙДЬБїТвНвµюјУ |
ХлґМІїО»·ўЙъ±д»ЇЈ¬ФміЙОЮР§»тОЈПХЈ»
ФміЙµз»чЙЛє¦ |
|
µзј«ХлБ¬ЅУЖчјРіЦБ¦І»Чг |
µзј«ХлБ¬ЅУЖчНСВ䴥ЕцЖдЛьХл»т»јХЯЙнМеЖдЛьІїО» |
ФміЙµз»чЙЛє¦Ј»
ЦОБЖОЮР§Ј» |
|
»ЇС§µДЈЁµзёЇКґЈ© |
µзХлТЗКдіцµДЦ±Бч·ЦБїК№Хл·ўЙъµз»ЇС§ёЇКґ |
·ўЙъ¶ПХл |
ХлµД¶ПМеІї·ЦБфФЪ»јХЯМеДЪ |
|
№¦ДЬЈЁІ»ККµ±КдіцЈ© |
РВµзіШµзС№№эёЯ»тідµзєуµзіШµзС№№эёЯ |
ІъЙъБЛёЯДЬБїµДВціеКдіц |
ФміЙµз»чЙЛє¦ |
К№УГґнОу
ЈЁІ»ЧсКШ№жФтЈ© |
І»ККµ±µДІЩЧчЛµГчєНІъЖ·О¬»¤±ЈСш |
·ЗЧЁТµТЅЙъК№УГЈ»Г»УРК№УГЛµГчКйєНјјКхЛµГчКйЈ¬»тЖдДЪИЭІ»И«ЎЈИзИ±ЙЩ±ШТЄµДѕЇёжЛµГчЎўИ±ЙЩПкПёµДК№УГ·Ѕ·ЁЎўИ±ЙЩ±ШТЄµДјјКхІОКэЎўИ±ЙЩµзВ·НјєНФЄЖчјюЗ嵥ЎўИ±ЙЩФЛКдєНЦьґж»·ѕіМхјюµДПЮЦЖЎўИ±ЙЩІъЖ·О¬»¤±ЈСшТЄЗуЎЈЙи±ёФЪµҐТ»№КХПЧґМ¬ПВФЛРР |
ІъЙъОЈПХЈ»ІъЖ·Лр»µ |
|
ІъЖ·ёЅјю(µзј«Хл)УРР§ЖЪ±кК¶І»ЗеОъ |
»јХЯЅУґҐµЅ№эЖЪУРѕъІъЖ· |
ёРИѕЈ¬СПЦШК±µјЦВЛАНц |
|
Т»ґОРФК№УГµДХл±»ТвНвµДЦШёґК№УГ |
±нГжСх»ЇµјµзРФІо |
ЦОБЖОЮР§»тБЖР§±дІо |
|
·ўЙъ¶ПХл |
ХлµД¶ПМеІї·ЦБфФЪ»јХЯМеДЪ |
|
Кдіц¶МВ·»тїЄВ· |
µзХлµДµзґМј¤±дЗї»тХЯГ»УР |
ФміЙґМј¤ДЬБї№эґу»тХЯЦОБЖОЮР§ |
|
К№УГК±ОґЅ«ЛщУРКдіц·щ¶ИЙи¶ЁОЄЧоРЎЦµЈ¬Кдіц·щ¶ИµчЅЪ№эїм№эґу |
Н»И»µДґуДЬБїКдіцЈЁґуВціе·щ¶ИЈ© |
ФміЙґМј¤ДЬБї№эґуЈ¬»јХЯІ»ККЈ¬·ўЙъµз»чЛрЙЛЎЈЙхЦБТэЖр»јХЯјЎИвЗїБТКХЛхЈ¬Тэ·ўМЫНґЎўНдХлЎў¶ПХл»тФОХлµИТвНв |
|
І»ХэИ·µДІъЖ·ЦьґжМхјюєНІъЖ·О¬»¤±ЈСшЎЈ |
ЖчјюАП»ЇЎўІїјюКЩГьЅµµН |
ІъЖ·КЩГьЅµµН |
|
№¦ДЬЙҐК§»т±д»µ |
µзј«ПЯАП»Ї |
ІъЖ·µДКдіцОЮ·ЁјУФШЦБХлЙП |
ОЮР§ЦОБЖ |
|
µзј«ХлБ¬ЅУЖчАП»Ї |
ОЮ·ЁУлХлїЙїїµјµзБ¬ЅУ |