|
|
| |
|
医疗器械软件产品注册审查指导原则(2022年修订版)
|
|
| [
2022-03-14 10:35 ] |
|
|
|
本指导原则旨在指导医疗器械注册申请人规范医疗器械软件生存周期过程和准备医疗器械软件注册申报资料,同时规范医疗器械软件的技术审评要求,为医疗器械软件、质量管理软件的体系核查提供参考。
本指导原则是对医疗器械软件的一般要求,注册申请人需根据产品特性和风险程度确定本指导原则具体内容的适用性,若不适用详述理由。注册申请人亦可采用其他符合法规要求的替代方法,但需提供详尽研究资料。
本指导原则是在现行法规、强制性标准体系以及当前科技能力、认知水平下制定的,随着法规、强制性标准体系的不断完善以及科技能力、认知水平的不断发展,本指导原则相关内容也将适时调整。
本指导原则作为注册申请人、审评人员和检查人员的指导性文件,不包括审评审批所涉及的行政事项,亦不作为法规强制执行,应在符合法规要求的前提下使用本指导原则。
本指导原则是数字医疗(Digital Health)指导原则体系的基础指导原则,亦是医疗器械软件的通用指导原则,其他含有或涉及软件的医疗器械指导原则可在本指导原则基础上进行有针对性的调整、修改和完善。
一、适用范围
本指导原则适用于医疗器械软件注册申报,包括第二、三类独立软件和含有软件组件的医疗器械(包括体外诊断医疗器械);适用于自研软件、现成软件的注册申报。
本指导原则也可用作医疗器械软件、质量管理软件的体系核查参考。
二、主要概念
(一)医疗器械软件
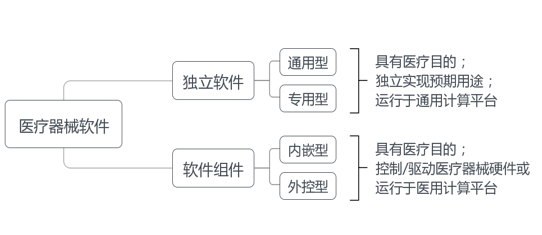
医疗器械软件包括本身即为医疗器械的软件或者医疗器械内含的软件,前者即医疗器械独立软件(简称独立软件),后者即医疗器械软件组件(简称软件组件),详见图1。
独立软件(SaMD)是指具有一个或多个医疗目的/用途,无需医疗器械硬件即可完成自身预期用途,运行于通用计算平台的软件[1]。通用计算平台满足信息技术设备安全要求(含电磁兼容),符合GB 4943.1、GB/T 9254等标准。
独立软件可分为通用型独立软件和专用型独立软件,前者通常基于通用数据接口与多个医疗器械联合使用,如医学图像处理软件、患者监护软件;后者基于通用、专用数据接口与特定医疗器械联合使用,可视为医疗器械附件,如动态心电数据分析软件、眼科显微镜图像处理软件。
软件组件(SiMD)是指具有一个或多个医疗目的/用途,控制/驱动医疗器械硬件或运行于医用计算平台的软件。医用计算平台满足医用电气设备(GB 9706系列)、实验室用电气设备(GB 4793系列)或有源植入式医疗器械(GB 16174系列)等安全要求(含电磁兼容);医用计算平台可与通用计算平台联合使用构成系统,整体视为医用计算平台。
软件组件可分为内嵌型软件组件和外控型软件组件,前者运行于医用计算平台,控制/驱动医疗器械硬件,如心电图机、脑电图机所含嵌入式软件(即固件);后者运行于通用计算平台,控制/驱动医疗器械硬件,如CT、MRI图像采集工作站软件[2]。
[1]详见IMDRF/SaMD WG/N10 FINAL: 2013。
[2]医用计算平台、软件组件可参考GB 9706.1-2020关于医用电气设备/系统、可编程医用电气设备/系统的定义和要求。
图1 医疗器械软件类型
独立软件作为医疗器械或医疗器械附件,通常单独注册,特殊情况可随医疗器械进行注册,此时虽不控制/驱动医疗器械硬件但从产品角度运行于医用计算平台,故视为软件组件,如专用型独立软件可作为附件随医疗器械进行注册。
软件组件作为医疗器械或医疗器械部件、附件的组成部分,不宜单独注册,需随医疗器械进行整体注册。
(二)系统软件、应用软件、中间件、支持软件
系统软件是指设计用于保障计算机系统正常运行的软件,如操作系统软件、虚拟机软件。应用软件是指设计用于实现计算机用户特定需求的软件,如浏览器软件、数据库软件、安全软件。中间件介于系统软件和应用软件之间,依赖于系统软件的支持,同时又为应用软件提供支持,如分布式计算平台软件。支持软件是指设计用于开发、测试其他软件的软件,如软件开发工具、软件测试工具。
医疗器械软件属于应用软件,其正常运行通常需要基于系统软件,或同时需要应用软件(含其他医疗器械软件)、中间件、支持软件的支持。
有些注册申请人会开发医用中间件用作医疗器械软件的公共支持平台,由于这些医用中间件是医疗器械软件正常运行所必需的,故作为医疗器械软件的组成部分予以考虑。
有些支持软件(如VTK、ITK)自带算法库,医疗器械软件开发过程已将算法库相关内容集成于自身内部,故医疗器械软件正常运行需要这些支持软件的支持。
本指导原则将医疗器械软件正常运行所必需的其他的医疗器械软件及医用中间件称为必备软件,而将其正常运行所必需的系统软件、通用应用软件、通用中间件、支持软件统称为外部软件环境。必备软件作为医疗器械软件单独注册,明确相互接口关系及技术特征即可。外部软件环境不含必备软件,亦非医疗器械软件。
(三)软件生存周期
软件生存周期(又称软件生命周期)是指软件系统从概念定义至停止使用的时间周期,包括软件开发策划、软件需求分析、软件设计、软件编码、软件测试、软件发布、软件部署、软件维护、软件停运等阶段。其中,从软件需求分析到软件发布的时间周期称为软件开发生存周期。
软件开发策划主要确定软件开发的目标和可行性。软件需求分析是将法规、标准、用户、产品等要求转换为软件需求规范/软件需求规格说明(SRS)。软件设计是通过设计活动将软件需求规范转换为软件设计规范/软件设计规格说明(SDS)。软件编码是通过编写源代码将软件设计规范转换为软件系统。软件测试是通过各类测试活动保证软件系统质量。软件发布是将软件系统予以产品定型。软件部署是指软件系统的交付、安装、设置和配置。软件维护是对软件系统发布后的更新需求予以实现。软件停运(即软件退市)是指终止软件系统的销售和售后服务,售后服务停止时间通常晚于停售时间。
软件生存周期模型是指一组包含过程、活动和任务的框架,跨越从软件需求分析到软件停运的软件生存周期过程,每个过程可细分为若干活动,每个活动又可细分为若干任务。其中,软件开发生存周期模型是软件生存周期模型的重要组成部分,常见模型包括瀑布模型、迭代模型、增量模型、V模型等。
敏捷开发是以人为核心、迭代与增量相结合的软件开发方法,常见软件开发生存周期模型包括SCRUM、极限编程等。敏捷开发秉承四条理念:人员互动胜于过程和工具,可用的软件胜于详尽的文档,客户合作胜于合同谈判,响应变化胜于遵循计划。因此,使用敏捷开发应兼顾质量管理体系相关要求,重点关注软件更新、文件与记录等控制要求。
医疗器械注册证申请人可结合软件的产品特点、风险程度以及质量管理体系要求,选择适宜的软件生存周期模型,参照相关国际、国家、行业标准建立相应软件生存周期过程。
(四)软件测试、软件验证、软件确认
软件测试是软件质量保证的基本措施,也是软件验证、软件确认的重要方法,从不同角度有不同分类方法。
从测试依据角度可分为黑盒测试和白盒测试。其中,黑盒测试是指基于输入与输出的测试,白盒测试是指基于源代码的测试,黑盒测试与白盒测试可组合使用,即灰盒测试。白盒测试根据是否运行源代码又可分为静态、动态分析/测试。
从测试进程角度可分为单元测试、集成测试、系统测试。其中,单元测试是对软件单元进行测试,通常采用白盒测试;集成测试是对软件项(由若干软件单元组成,即软件模块)进行测试,白盒测试、黑盒测试、灰盒测试相结合;系统测试是对软件系统(由若干软件项组成)进行测试,通常采用黑盒测试,其从测试内容角度又可分为功能测试、性能测试、并发测试、压力测试、接口测试、内存测试、兼容性测试、用户界面测试、安装卸载测试、安全测试等。
从测试实施方角度可分为内部测试、用户测试、第三方测试。其中,内部测试是指注册申请人实施的测试,包括单元测试、集成测试、系统测试,白盒测试、黑盒测试、灰盒测试相结合;用户测试是指预期用户在真实或模拟使用场景对软件系统进行测试,采用黑盒测试;第三方测试是指第三方机构对软件系统进行测试,通常采用黑盒测试。
回归测试是指用于确定软件更新没有产生不良影响且未引入风险不可接受新缺陷的软件测试。回归测试需根据软件更新的类型、内容和程度,开展与之相适宜的单元测试、集成测试、系统测试、用户测试、第三方测试等测试活动。
软件验证是指通过提供客观证据认定软件开发、软件维护某一阶段的输出满足输入要求。软件验证包括源代码审核、静态和动态分析/测试、单元测试、集成测试、系统测试、设计评审等系列活动,是软件确认的基础。
软件确认是指通过提供客观证据认定软件满足用户需求和预期用途。软件确认是基于过程控制的设计确认,包括用户测试、临床评价、设计评审等系列活动,即要保证软件满足用户需求和预期用途,又要确保软件已知剩余缺陷的风险均可接受。
医疗器械产品注册申请人需结合软件的产品特点、风险程度考虑相应软件测试要求,明确语句、判定、条件、路径等测试覆盖率要求,以保证软件验证、软件确认的质量。全部源代码均应测试,可结合白盒测试、黑盒测试、灰盒测试等方法予以实现。
(五)软件可追溯性分析
软件可追溯性分析作为软件验证、软件确认的重要活动之一,是指追踪软件需求、软件设计、源代码、软件测试、软件风险管理之间的关系,分析已识别关系的正确性、一致性、完整性、准确性。
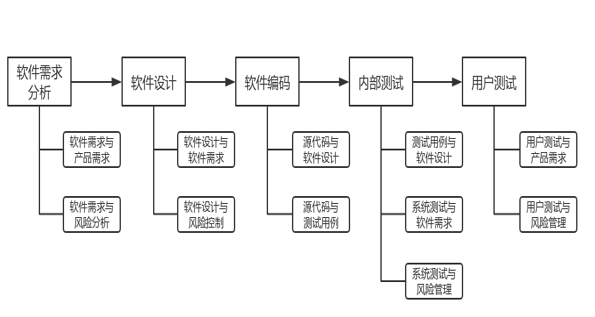
软件生存周期过程均需开展可追溯性分析活动,详见图2。软件需求分析阶段追溯分析软件需求与产品需求、软件需求与风险分析的关系。软件设计阶段追溯分析软件设计与软件需求、软件设计与风险控制的关系。软件编码阶段追溯分析源代码与软件设计、源代码与测试用例的关系。内部测试阶段追溯分析单元测试、集成测试、系统测试各级测试用例与软件设计,系统测试与软件需求,系统测试与风险管理的关系。用户测试阶段追溯分析用户测试与产品需求、用户测试与风险管理的关系。软件更新亦需开展与之相适宜的软件可追溯性分析活动。
图2 软件可追溯性分析
注册申请人应建立软件可追溯性分析过程,规范软件可追溯性分析相关活动要求,以保证软件验证、软件确认的质量。考虑到行业实际情况,源代码追溯分析活动追溯至软件单元(列明名称)即可。
(六)软件更新
1. 主要概念
软件更新是指注册申请人在软件生存周期全过程对软件所做的任一修改,亦称软件变更、软件维护。软件维护通常与软件更新含义相同,但可特指发布后软件更新。
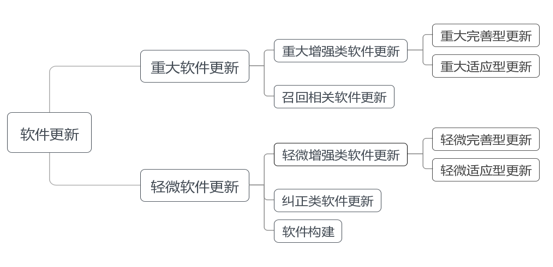
软件更新从不同角度出发有不同分类方法。从更新结果角度可分为重大更新和轻微更新,重大更新是指影响到医疗器械安全性或有效性的软件更新,反之即为轻微更新。
从更新内容角度可分为增强类更新和纠正类更新(即软件缺陷修复)。增强类更新又可分为完善型更新和适应型更新,其中完善型更新是指改变功能、性能、接口等软件属性的软件更新,适应型更新是指适应新运行环境的软件更新;其从更新结果角度又可分为重大增强类更新、轻微增强类更新。纠正类更新又可分为修正型更新和预防型更新,其中修正型更新是指修复软件存在且已造成运行故障缺陷的软件更新,预防型更新是指修复软件存在但尚未造成运行故障缺陷的软件更新;其通常属于轻微更新,除非影响到医疗器械安全性或有效性。
本指导原则关注医疗器械软件的安全性和有效性,将软件更新分为:
(1)重大软件更新:影响到医疗器械安全性或有效性的增强类更新,即重大增强类软件更新,应申请变更注册。
(2)轻微软件更新:不影响医疗器械安全性与有效性的增强类更新、纠正类更新,包括轻微增强类软件更新、纠正类软件更新,通过质量管理体系进行控制,无需申请变更注册,待下次变更注册时提交相应注册申报资料。
此外,软件构建是指软件编译生成一个工作版本,符合软件更新定义,通过质量管理体系进行控制,注册申报资料要求与纠正类软件更新相同。召回相关软件更新包括软件更新导致召回、召回措施所用软件更新,无论增强类更新还是纠正类更新均属于重大软件更新,按照医疗器械召回相关法规要求处理,不属于本指导原则讨论范畴。
软件更新遵循风险从高原则,即同时发生重大、轻微软件更新按重大软件更新处理,同时发生增强类、纠正类软件更新按增强类软件更新处理。软件更新需考虑引入回滚机制,以保证医疗业务的连续性,特别是对高风险软件。
软件重新开发即注册医疗器械申请人弃用原有软件而开发新软件,不属于软件更新范畴,按初次发布处理。
2. 重大软件更新判定原则
软件更新若影响到医疗器械的预期用途、使用场景或核心功能原则上均属于重大软件更新,其判定原则包括但不限于:
(1)完善型软件更新:若影响到用户决策(含决策能力、决策结果、决策流程、用户行动)或人员(含患者、用户、其他相关人员)安全则属于重大软件更新,如软件的输入输出数据类型、体系结构、用户界面关系、物理拓扑、核心算法、核心功能、诊疗流程或预期用途等发生改变,软件系统、高风险软件项/软件单元进行代码重构,安全性级别改变,调整报警方式等;而运算效率单纯提高、诊疗流程或工作语言可配置化(即用户可保留原有诊疗流程或工作语言)、用户界面文字性修改、中低风险软件项/软件单元的代码重构等情况通常不视为重大软件更新,除非影响到医疗器械的安全性或有效性。
(2)适应型软件更新:若软件运行环境跨越互不兼容的计算平台(含硬件配置、外部软件环境、必备软件、网络条件)则属于重大软件更新,如预期运行的操作系统软件由Windows变为iOS,更换浏览器内核、必备软件,网络条件由局域网变为广域网,计算平台由通用计算平台变为医用计算平台等;预期运行的系统软件、支持软件、通用中间件的兼容性版本更新、补丁更新通常不视为重大软件更新,除非影响到医疗器械的安全性或有效性。
综上,医疗器械软件更新类型如图3所示。
图3 医疗器械软件更新类型
重大软件更新的范围会随着认知水平与技术能力的提高、不良事件与召回情况的分析进行动态调整。
(七)软件版本
软件没有物理实体,只能通过状态标识进行软件更新管理,从而保证软件质量。软件版本使用不同字段来区分软件更新类型,进而标识软件状态,因此软件版本与软件是一一对应的表里关系,亦是软件标识不可或缺的组成部分。
软件版本可分为软件发布版本和软件完整版本[1],其中软件发布版本仅体现重大软件更新,即只限于重大增强类软件更新,其改变意味着发生重大软件更新,反之亦然;软件完整版本体现重大、轻微软件更新的全部类型,包括重大增强类软件更新、轻微增强类软件更新、纠正类软件更新、软件构建(若适用),其不同字段的改变意味发生不同类型的软件更新,反之亦然。
软件发布版本发生改变表示软件发生重大更新,应申请变更注册,软件完整版本发生改变但软件发布版本未变表示软件仅发生轻微更新,此时通过质量管理体系进行控制,无需申请变更注册。例如,软件版本命名规则为X.Y.Z.B,其中X表示重大增强类软件更新,Y表示轻微增强类软件更新,Z表示纠正类软件更新,B表示软件构建,则软件发布版本为X,软件完整版本为X.Y.Z.B,此时X改变应申请变更注册,而Y、Z、B改变但X未变则无需申请变更注册。
注册申请人应综合考虑软件产品特点、质量管理体系要求、合规性等因素制定软件版本命名规则并予以记录,明确字段的位数、范围、含义,涵盖软件更新全部类型,字段含义明确且无歧义无矛盾,能够区分重大软件更新和轻微软件更新,保证软件更新的版本变更符合软件版本命名规则要求。同时,考虑医疗器械网络安全、人工智能医疗器械等指导原则的要求。
软件版本命名规则同样遵循风险从高原则,即某字段同时表示重大软件更新和轻微软件更新,则该字段按重大软件更新处理,并作为软件发布版本的组成部分。
有用户界面的软件可在登录界面、主界面、“关于”或“帮助”等界面体现软件发布版本、软件完整版本,两个版本均需体现但无需每个界面同时体现。无用户界面的软件需提供获取软件完整版本的方法,以明确软件版本信息[2]。
产品技术要求注明软件发布版本、软件版本命名规则,其中软件版本命名规则需与质量管理体系保持一致。检测报告提供软件版本界面照片或列明软件版本信息,有用户界面的软件体现软件发布版本、软件完整版本,无用户界面的软件体现软件完整版本。说明书注明软件发布版本。
软件模块(含医用中间件)若单独进行版本控制,亦需满足版本控制要求,并明确与软件系统版本控制的关系。
(八)软件算法、软件功能、软件用途
软件算法是软件功能的基础,二者是多对多的关系,即一个软件算法可供一个或多个软件功能使用,一个软件功能可含一个或多个软件算法。同理,软件功能和软件用途的关系亦是如此。
从用户角度出发,软件算法是内在不可见的,软件功能和软件用途是外在可见的,考虑到软件功能是软件算法、软件用途的联系纽带,故以软件功能作为软件安全有效性评价主线。例如,区域生长算法可实现图像分割功能,图像分割功能可用于病变轮廓标识。
软件功能从不同角度出发有不同分类方法。从重要性角度可分为核心功能和非核心功能,其中核心功能是指软件在预期使用场景完成预期用途所必需的功能,反之即为非核心功能。
从技术特征角度大体上可分为处理功能、控制功能、安全功能,其中处理功能是指加工医疗器械数据(即医疗器械产生的用于医疗用途的客观数据)或基于模型计算(如辐射剂量模型、药代模型)的功能,控制功能是指控制/驱动医疗器械硬件运行的功能,安全功能是指保证医疗器械安全性的功能。
处理功能从数据流角度又可分为前处理功能和后处理功能,前者是指采集人体解剖、生理信息生成医疗器械数据过程的处理功能,如滤波、降噪、校正、重建等功能;后者是指利用医疗器械数据生成诊疗信息或进行医疗干预过程的处理功能,如平移、缩放、旋转、滤波、测量、分割、融合、三维重建、治疗计划制定、药代模型计算、基因测序、分析等功能。后处理功能从复杂性角度又可分为简单功能和复杂功能,前者是指对现有医疗信息进行操作而非生成新医疗信息的功能,如平移、缩放、旋转等功能;后者是指生成新医疗信息的功能,如滤波、测量、分割、融合、三维重建、治疗计划制定、药代模型计算、基因测序、分析等功能。
独立软件的功能均为后处理功能,软件组件的功能以控制功能、前处理功能为主,兼具后处理功能。考虑到控制功能和前处理功能通常与医疗器械硬件产品共同评价,故处理功能若无明示均指后处理功能;同时,考虑到测量、模型计算、分析等功能具有特殊性,通常情况下与处理功能并列表述。
从监管范围角度可分为医疗器械功能和非医疗器械功能,其中医疗器械功能是指可用作医疗器械界定依据的软件功能,如医学图像、生理参数、体外诊断等数据的测量、处理、模型计算、分析等功能;反之即为非医疗器械功能,如收费计价、行政办公等功能,不属于监管对象;实现医疗器械功能所必需的患者信息管理功能属于医疗器械功能。二者尽量通过模块化设计予以拆分,若在技术上无法拆分需将非医疗器械功能作为医疗器械软件的组成部分予以整体考虑。
软件算法从重要性角度可分为核心算法和非核心算法,其中核心算法是指实现软件核心功能所必需的算法,反之即为非核心算法。从复杂性角度可分为简单算法和复杂算法,前者原理简单明确或基于成熟公式,后者通常基于模型研究,存在诸多假设条件且影响因素较多,同时二者在算法规模、参数数量、运算速度等方面亦存在差异。从可解释性角度可分为白盒算法和黑盒算法,前者可与现有知识建立关联,后者难与现有知识建立关联,前者可解释性优于后者。
软件用途通常可分为辅助决策和非辅助决策,其中辅助决策是指通过提供诊疗活动建议辅助用户(如医务人员、患者)进行医疗决策,如病灶特征识别、病灶性质判定、用药指导、制定治疗计划等,相当于用户的“助手”;反之,仅提供医疗参考信息而不进行医疗决策即为非辅助决策,包括流程优化、诊疗驱动,前者如诊疗流程简化等,后者如测量、分割、三维重建等,相当于用户的“工具”。同时,辅助决策和非辅助决策从实时性角度均可细分为实时和非实时,前者风险通常高于后者。故软件功能从软件用途角度又可分为辅助决策类功能和非辅助决策类功能、实时功能和非实时功能。
软件算法、软件功能、软件用途从成熟度角度均可分为成熟和全新两种类型,其中成熟是指安全有效性已在医疗实践中得到充分证实的情形,全新是指未上市或安全有效性尚未在医疗实践中得到充分证实的情形[3]。同理,软件亦可分为全新软件和成熟软件,软件的算法、功能、用途若有一项为全新类型则属于全新软件,反之属于成熟软件。因全新软件的潜在未知风险多于成熟软件,故本指导原则以核心功能、核心算法为基础,重点关注全新软件的安全有效性。
三、基本原则
(一)基于软件特性
随着信息通讯技术的迅猛发展,软件在医疗器械中的应用日益普遍,作用日趋重要,开发形式灵活多变,新技术层出不穷。但随之而来的质量问题也日益增多,医疗器械召回数据表明软件相关召回数量持续增加,明显高于同期医疗器械整体水平,同时软件失效导致患者死亡或严重伤害的召回事件也屡见不鲜,因此软件质量问题的严重性不容忽视,需要基于软件特性加强软件质量保证工作。
软件没有物理实体,在开发和使用过程中人为因素影响无处不在,软件测试由于时间和成本的限制不能穷尽所有情况,所以软件缺陷无法避免。同时,软件更新频繁且迅速,细微更新可能导致严重后果,并存在累积效应和退化问题(即每修复若干个缺陷就会产生一个新缺陷),所以软件缺陷无法根除。因此,软件缺陷可视为软件的固有属性之一,这也是软件质量问题较为突出的根源所在。
软件与硬件存在诸多差异:硬件是物理实体,软件是逻辑关系;硬件变更周期较长,软件更新容易、快速且频繁;硬件有磨损问题,软件虽无磨损但有累积效应和退化问题;硬件质量取决于设计开发和生产,软件质量取决于设计开发,与生产基本无关;硬件失效先有征兆再发生,软件失效往往没有征兆突然发生,软件失效率远高于硬件;硬件部件可以标准化,软件模块的标准化较为复杂;细微变更对硬件影响有限,但对软件影响可能严重;硬件检验可基本保证质量,软件测试不足以保证质量。这些差异使得传统硬件质量控制方法对于软件质量保证往往达不到预期效果。
鉴于软件特性,只有综合考虑风险管理、质量管理和软件工程的要求才能保证软件的安全有效性。注册申请人需基于软件风险程度,采用良好软件工程实践完善质量管理体系,针对算法、接口、更新、异常处理等软件召回主要原因,尽早、重点、全面开展软件质量保证工作。
(二)风险导向
综合考虑软件使用的普遍性、监管资源的有限性和风险分级管理导向,软件风险程度不同,其生存周期质控要求和注册申报资料要求亦不同。
软件风险程度采用软件安全性级别进行表述,软件安全性级别越高,生存周期质控要求越严格,注册申报资料也越详尽。软件安全性级别基于软件风险程度分为轻微、中等、严重三个级别[4],其中轻微级别即软件不可能产生伤害,中等级别即软件可能直接或间接产生轻微(不严重)伤害,严重级别即软件可能直接或间接产生严重伤害或导致死亡。
软件安全性级别可结合软件的预期用途、使用场景、核心功能进行综合判定[5]。其中,预期用途主要考虑软件的用途类型(如治疗、诊断、监护、筛查)、重要程度(如重要作用、参考作用、补充作用)、紧迫程度(如危重情形、严重情形、普通情形)、成熟程度(成熟、全新)等因素,使用场景主要考虑软件的使用场所(如门诊、手术、住院、急救、家庭、转运、公共场所)、疾病特征(如严重性、紧迫性、传染性)、适用人群(如成人、儿童、老人、孕妇)、目标用户(如医务人员、患者)等因素,核心功能主要考虑软件的功能类型(如重要程度、技术特征、复杂程度、成熟程度)、核心算法(如重要程度、复杂程度、可解释性、成熟程度)、输入输出(输入数据如医学图像、生理参数、体外诊断等数据,输出结果如处理、测量、分析等结果)、接口(如应用程序接口(API)、数据接口、产品接口)等因素。
软件安全性级别也可根据风险管理所确定的风险等级进行判定,软件安全性级别与风险等级的分级可以不同,但二者存在对应关系,因此可根据风险等级来判定软件安全性级别,但应在采取风险控制措施之前进行判定,后续可通过外部风险控制措施(含软件措施、硬件措施)降低初始软件安全性级别。
软件风险管理需要注意:软件本身不是危险,但可能会引发危险情况;软件失效虽表现为随机性失效但实为系统性失效,同时软件失效所致危险的发生概率难以统计,故软件风险程度基于伤害严重度并可结合危险情况所致伤害的概率进行判定;软件组件需与所属医疗器械整体开展风险管理工作。
软件安全性级别亦可参考已上市同类医疗器械软件的不良事件和召回情况进行判定,即已上市同类医疗器械软件若发生严重不良事件或一级召回属于严重级别,发生不良事件或二级召回属于中等级别,未发生不良事件且仅发生三级召回或无召回属于轻微级别。
注册申请人应结合质量管理体系要求建立相应软件生存周期过程,并开展与软件安全性级别相匹配的软件质量保证工作。同时,基于软件安全性级别提交相应注册申报资料,注册申报资料均来源于软件生存周期过程所形成的文档。
独立软件和软件组件虽然存在技术差异,但生存周期过程质控原则和要求均相同,故二者注册申报资料要求基本一致,具体内容略有差异,详见第八章。
(三)全生命周期质控
由于软件本身的复杂性和软件测试的局限性,软件开发过程的质量保证活动不足以保证软件的安全有效性,因此应在医疗器械全生命周期中考虑软件质控要求,并将软件风险管理、软件配置管理、软件缺陷管理、软件可追溯性分析贯穿于医疗器械生命周期全程。
上市前开展充分有效的软件验证与确认活动,识别软件可预见的风险并将其降至可接受水平。上市后继续开展软件质量保证工作,结合用户投诉、不良事件和召回等情况识别前期未预见的风险,并采取必要措施保证软件质量;同时,基于软件更新需求的评估,实施软件更新活动以满足用户新需求,并开展与之相适宜的软件验证与确认活动,以保证软件更新质量;此外,软件停运考虑用户告知与后续服务、数据迁移、患者数据与隐私保护等要求。
总之,数字医疗技术处于快速发展阶段,新技术层出不穷,对医疗器械行业的影响日益深远,其监管问题亟需加强研究。无论何种数字医疗新技术,软件作为其技术基础,仍可遵循上述三条基本原则,开展相应安全有效性评价工作。
四、现成软件
(一)主要概念
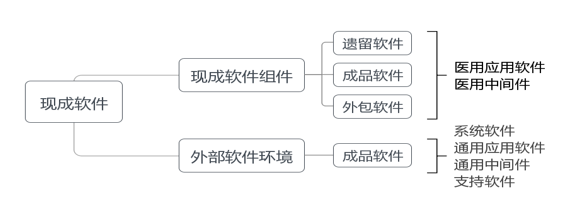
医疗器械注册申请人进行完整生存周期控制的软件称为自研软件(若无明示简称为软件),反之注册申请人未进行(含无法证明)完整生存周期控制的软件称为现成软件,包括遗留软件、成品软件、外包软件。其中,遗留软件是指注册申请人以前开发但现在不能得到足够开发记录的软件,即注册申请人无法证明其进行完整生存周期控制的软件,涉及应用软件、中间件。成品软件是指第三方开发的且通常可得到的软件,即注册申请人未进行完整生存周期控制的软件,涉及系统软件、应用软件、中间件、支持软件,如开源/闭源软件、免费/付费软件等。外包软件是指注册申请人委托第三方开发的软件,即注册申请人仅进行部分生存周期控制的软件,涉及应用软件、中间件。
现成软件与医疗器械软件的关系可分为两类。一类是现成软件作为医疗器械软件的组成部分,即现成软件组件,包括遗留软件、成品软件、外包软件,涉及医用应用软件、医用中间件,属于监管对象;无论是否设计用于医疗用途,现成软件组件作为医疗器械软件的组成部分,其功能均属于医疗器械功能,原因在于若为非医疗器械功能则应从医疗器械软件中直接删除。另一类是现成软件作为医疗器械软件运行环境的组成部分,即外部软件环境,主要为成品软件,涉及系统软件、通用应用软件、通用中间件、支持软件,虽非监管对象,但需从风险管理角度考虑其对医疗器械软件的影响。
综上,现成软件类型详见图4。
[1]从医疗器械唯一标识(UDI)角度,软件完整版本属于生产标识(PI)的组成部分。
[2] 详见IMDRF/UDI WG/N7 FINAL: 2013。
[3]详见IMDRF/SaMD WG/N41 FINAL: 2017。
[4]轻微级别、中等级别、严重级别分别与YY/T 0664所定义的A级、B级、C级相对应。
[5]详见IMDRF/SaMD WG/N12 FINAL: 2014。
图4 现成软件类型
医疗器械软件除部分内嵌型软件组件外,均需外部软件环境的支持方能正常运行。同时,医疗器械软件既可自研软件和现成软件组件相结合,部分使用现成软件组件,即部分使用方式;又可全部使用现成软件组件,即全部使用方式。因此,现成软件的使用具有普遍性、灵活性、复杂性等特点。
由于现成软件通常并非设计用于医疗,特别是外部软件环境,未必能够满足医疗器械相关要求,未用功能可能通过耦合关系对医疗器械软件产生不利影响,而且注册申请人未对现成软件进行完整生存周期控制,因此使用现成软件的风险相对较高。此外,现成软件组件直接实现医疗用途,风险相对更高。
注册申请人使用现成软件应保证医疗器械软件的安全有效性,成品软件和外部软件环境的开发商作为医疗器械供应商并不承担注册申请人相关责任。因此,注册申请人需结合现成软件的类型、特点以及业界使用情况,区分现成软件组件和外部软件环境,综合考虑现成软件的使用广泛度、技术成熟度、售后支持程度以及功能、性能、兼容性、易用性、可靠性、维护性、可移植性、网络安全等要求,采用基于风险的全生命周期管理方法进行质控,特别是在采购、设计开发、上市后监测等方面,同时开展差距分析,必要时采取补救措施以保证安全有效性。
由于自研软件与现成软件、现成软件组件与外部软件环境、部分使用方式与全部使用方式的质控要求均存在差异,故相应注册申报资料均有所不同,具体要求详见第八章。此外,医疗器械软件可同时使用多个版本、多个、多种的现成软件,需进行整体考量并提供相应医疗器械软件注册申报资料。
(二)现成软件通用考量
1. 现成软件组件
现成软件组件的软件更新、软件版本相关要求与自研软件基本相同,同样遵循风险从高原则。
现成软件组件若发生重大软件更新亦应申请变更注册,若发生轻微软件更新通过质量管理体系进行控制,无需申请变更注册。
注册申请人应根据质量管理体系要求,制定现成软件组件的版本命名规则,亦需考虑合规性要求。若现成软件组件开发商的软件版本命名规则满足合规性要求,可直接采用。
2. 外部软件环境
医疗器械软件与外部软件环境存在耦合关系,需整体考量。
从医疗器械软件角度,软件安全性级别判定需考虑外部软件环境失效的影响,软件需求规范文档、软件测试计划列明外部软件环境的基本情况,软件测试报告列明外部软件环境所含各现成软件的名称、完整版本、补丁版本。软件验证与确认基于外部软件环境所含全部现成软件予以实施,若使用同一现成软件的多个版本,则每个版本均需进行软件验证与确认。根据风险控制要求,医疗器械软件必要时需具备外部软件环境自检功能,以确保自身能够正常运行。同理,说明书必要时需明确外部软件环境所含全部现成软件的交付、安装、设置、配置、更新以及使用限制、警示提示等内容。
从外部软件环境角度,自身风险相对较低,由于与医疗器械软件相互耦合,故其安全性级别与医疗器械软件的安全性级别相同,注册申报资料详尽程度亦取决于其安全性级别。
外部软件环境更新对于医疗器械软件而言属于适应型软件更新,包括产品更新(即更换外部软件环境所含现成软件)、版本更新、补丁更新等情况。外部软件环境更新情况不同,对于医疗器械软件的影响亦不同,通常补丁更新、与医疗器械软件兼容的版本更新属于轻微软件更新,而产品更新、为解决与医疗器械软件不兼容问题的版本更新属于重大软件更新,同样遵循风险从高原则。因此,注册申请人需依据相应流程开展外部软件环境更新的影响评估工作。
五、质量管理软件
(一)主要概念
质量管理软件是指医疗器械质量管理所用的应用软件,非医疗器械软件,无需申报注册。质量管理软件与医疗器械软件在技术层面具有相同之处,故可参照医疗器械软件相关要求考虑质量管理软件的确认要求。
质量管理软件参照医疗器械软件可分为类独立软件和类软件组件,前者包括设计开发所用软件、流程管理所用软件等,后者包括生产设备所含软件、检验设备所含软件等。
质量管理软件多数通过采购获得,特别是类软件组件,可视为成品软件,主要采用全部使用方式,若二次开发则属于部分使用方式;少数自行研发,主要是类独立软件,可视为自研软件。因此,质量管理软件确认通常可参照成品软件、自研软件的确认要求。
质量管理软件亦需外部软件环境(含系统软件、通用应用软件、通用中间件、支持软件)的支持方能正常运行,故其确认亦需考虑外部软件环境的评估要求。
(二)质量管理软件确认考量
质量管理软件确认以软件功能为基础,综合考虑需求分析、验收测试、维护计划等要求。其中,需求分析考虑软件的功能、性能、接口等要求,评估候选软件以确定目标对象。验收测试基于外部软件环境予以实施,确保软件能够满足使用需求,而且软件已知剩余缺陷、未用软件功能对于医疗器械质量的影响均可接受。维护计划考虑软件更新相关要求,特别是纠正类软件更新(即软件缺陷修复)的维护方案。
质量管理软件外部软件环境的评估要求可参照自研软件相应要求。质量管理软件更新应重新进行软件确认,其外部软件环境更新亦需开展影响评估工作。
六、医疗器械软件生存周期过程
软件生存周期过程主要包括软件开发过程、软件维护过程、软件风险管理过程、软件配置管理过程、软件缺陷管理过程。
软件开发过程包括软件开发策划、软件需求分析、软件设计、软件编码、软件验证、软件确认、软件发布等活动。
软件维护过程适用于发布后增强类软件更新,包括软件更新需求评估、软件更新策划、软件更新实施、软件验证、软件确认、软件发布、用户告知等活动。
软件风险管理过程包括风险分析、风险评价、风险控制、综合剩余风险评价等活动,基于医疗器械风险管理过程予以实施,可采用医疗器械常用风险管理方法。
软件配置管理过程包括配置项识别、更改控制、配置状态记录等活动,基于软件版本命名规则进行软件版本控制,可使用配置管理工具或常用办公软件予以实施。软件版本命名规则明确字段的位数、范围、含义,字段含义明确且无歧义无矛盾,涵盖自研软件、现成软件、网络安全的全部软件更新类型,能够区分重大软件更新和轻微软件更新,保证软件更新的版本变更符合软件版本命名规则要求[1]。
软件缺陷管理过程适用于发布前和发布后纠正类软件更新,包括软件缺陷评估、软件缺陷修复、回归测试等活动,可使用缺陷管理工具或常用办公软件予以实施。
软件开发过程、软件维护过程是前后关系,软件风险管理过程、软件配置管理过程、软件缺陷管理过程贯穿于软件开发过程、软件维护过程。
同时,软件可追溯性分析也是软件生存周期过程的重要过程之一,同样贯穿于软件开发过程、软件维护过程,可使用可追溯性分析工具或常用办公软件予以实施。
此外,软件生存周期过程亦需考虑现成软件、网络安全相关要求。现成软件考虑采购控制、设计开发控制等要求,使用外包软件需与供应商签订质量协议,使用遗留软件需评估现有文件、上市后使用问题(含不良事件、召回)等情况,使用开源软件需遵循相应开源许可协议。网络安全设计开发与软件设计开发相融合,可基于网络安全能力建设要求予以实施,并考虑网络安全事件应急响应要求。
软件生存周期过程质量保证活动要求应与软件安全性级别相匹配,软件风险程度越高,其质控要求越严格。
敏捷开发具有特殊性,还需加强软件更新、文件与记录等控制要求。
软件生存周期过程(包括现成软件、网络安全)的具体要求详见医疗器械独立软件生产质量管理规范及其现场检查指导原则[2]。
七、技术考量
(一)注册单元与检测单元
1. 注册单元划分原则
(1)独立软件
独立软件注册单元以管理类别、预期用途、功能模块作为划分原则。
不同管理类别的独立软件作为不同注册单元,若在技术上无法拆分可作为一个注册单元并按照较高管理类别申报注册。
不同预期用途的独立软件作为不同注册单元,按照预期用途可分为辅助决策类和非辅助决策类,每类又可细分为治疗、诊断、监护、筛查等情形。
对于功能庞大复杂的独立软件,依据功能模块的类型和数量划分注册单元,每个注册单元所含功能模块数量需适中。按照功能模块类型可分为平台功能软件和特定功能软件[3],其中平台功能软件作为软件平台提供基本功能和共用功能,支持多模态数据(如医学图像、生理参数、体外诊断等数据);特定功能软件运行于平台功能软件并提供特定功能,支持单模态数据或者使用多模态数据实现某一特定预期用途。
例如,某PACS包含数十个单独的功能模块,并含有辅助决策类功能模块,需拆分为一个平台功能软件和多个特定功能软件,其中辅助决策类功能模块单独作为一个注册单元。
(2)软件组件
软件组件注册单元与所属医疗器械相同,有软件组件和无软件组件的医疗器械作为不同注册单元。专用型独立软件视为软件组件的注册单元与软件组件相同。
2. 检测单元划分原则
检测单元是指同一注册单元内用于检测的代表产品。
(1)独立软件
独立软件检测单元原则上与注册单元相同,但若有多个运行环境或多个发布版本,则每个互不兼容的运行环境(含云计算)或每个互不涵盖的发布版本均需作为一个检测单元。
若软件核心功能相同但核心算法类型不同,则每类核心算法所对应的核心功能均需检测(检测对象为核心功能而非核心算法)。例如,图像分割功能所用核心算法含常规图像处理算法和人工智能算法,基于这两类算法的图像分割功能均需检测。
(2)软件组件
软件组件检测单元原则上与所属医疗器械相同,但医疗器械若包含多个软件组件或多个发布版本的软件组件,则每个软件组件或每个发布版本的软件组件均需作为一个检测单元,除非检测单元能够完整覆盖注册单元全部情况。同理,若软件核心功能相同但核心算法类型不同,则每类核心算法所对应的核心功能均需检测。
专用型独立软件视为软件组件的检测单元原则上与软件组件相同,但若有多个运行环境,则每个互不兼容的运行环境(含云计算)均需作为一个检测单元。
(二)临床评价基本原则
本指导原则仅明确医疗器械软件临床评价的基本原则[4],具体要求详见医疗器械临床评价相关指导原则。
1. 独立软件
独立软件通常基于软件功能进行临床评价,必要时可基于软件算法进行临床评价。临床评价需结合独立软件的预期用途和成熟度予以综合考虑,可选择已上市医疗器械所含同类软件功能进行同品种医疗器械比对。
非辅助决策类软件功能基于核心功能进行同品种医疗器械比对。全新的核心算法、核心功能、预期用途原则上均需开展临床评价。简单操作类、单纯流程优化类软件功能可通过非临床证据予以评价。
辅助决策类软件功能基于核心算法进行同品种医疗器械比对,所选同品种医疗器械的临床证据原则上需基于临床试验(含回顾性研究,下同)。全新的核心算法、核心功能、预期用途原则上均需开展临床试验。临床试验若采用阳性对照设计,可选择预期用途相同且核心算法或核心功能等同的独立软件进行对照。
2. 软件组件
软件组件控制功能、前处理功能的临床评价通常与所属医疗器械进行整体评价。后处理功能的临床评价可参照独立软件要求,亦可随所属医疗器械进行整体评价。
专用型独立软件视为软件组件的临床评价与软件组件后处理功能要求相同。
(三)网络安全
医疗器械网络安全需从网络安全角度综合考虑信息安全和数据安全。医疗器械软件若具备电子数据交换、远程访问与控制、用户访问三种功能当中一种及以上功能,均需考虑网络安全问题,具体要求详见医疗器械网络安全相关指导原则。
(四)云计算[5]
云计算在医疗器械行业的应用日益增多,虽然其具有降低信息化成本、减少重复建设、提高资源利用率、增加业务灵活性、提升服务专业性等优势,但是也存在着用户对数据控制能力减弱、服务可持续性降低、数据所有权面临挑战、数据保护困难、数据残留难以处理、用户与云服务商责任不清、产生司法管辖权问题、面临网络安全威胁等风险,因此注册申请人需权衡使用云计算的受益和风险。
云计算视为现成软件,云服务商视为医疗器械供应商,因此,注册申请人可参照现成软件和医疗器械供应商相关要求,考虑云计算的需求分析、风险管理、验证与确认、维护计划等活动要求。
需求分析需考虑云计算的技术特征、云服务商的选择问题。云计算技术特征包括服务模式、部署模式、配置情况、核心功能、数据接口、网络安全保证等方面,其中服务模式分为软件即服务(SaaS)、平台即服务(PaaS)、基础设施即服务(IaaS),部署模式分为私有云、公有云、混合云,配置情况包括计算资源、配套软件功能等要求,核心功能包括数据存储、数据处理、数据分析等功能,数据接口考虑传输协议、存储格式等要求,网络安全保证考虑数据匿名、数据加密、数据传输校验、安全配置等技术措施。同时,基于云计算的服务资质、服务协议等因素考虑云服务商选择问题,云计算服务协议需明确网络安全保证、患者数据与隐私保护等责任承担要求。
风险管理基于云计算的核心功能、数据接口、网络安全保证予以实施。验证与确认基于云计算环境开展医疗器械软件的验证与确认工作,确保云计算满足使用要求,且已知剩余缺陷的风险均可接受。维护计划考虑云计算更新的维护方案,重新开展医疗器械软件的验证与确认工作,明确云计算服务终止的无损数据迁移方案。
若使用云计算,注册申请人需在自研软件研究报告、外部软件环境评估报告相关条款中予以体现,具体要求详见第八章。若自建云计算平台,注册申请人应遵循云服务商相关规定,参照自研软件要求提交相应研究资料。
(五)移动计算
医疗器械软件若运行于供个人使用的移动计算终端(含医用终端、通用终端),则属于移动医疗器械。此时需综合考虑移动计算终端的技术特征及其风险,具体要求详见移动医疗器械相关指导原则。
(六)人工智能
医疗器械软件若使用人工智能技术实现其预期用途(即医疗用途),则属于人工智能医疗器械。此时需综合考虑人工智能算法的技术特征及其风险,具体要求详见人工智能医疗器械相关指导原则。
(七)人因与可用性
建议加强医疗器械软件用户界面的人因设计以提升可用性,将用户错误使用的风险降至可接受水平,具体要求详见医疗器械人因设计相关指导原则。
(八)互操作性
互操作性(又称互用性)是指医疗器械与其他医疗器械或通用设备通过电子接口交换并使用信息的能力。其中电子接口包含硬件接口和软件接口,信息包括但不限于医学图像、生理参数、体外诊断等数据和控制指令。
互操作性重点关注医疗器械软件的接口设计及其风险,接口包括内部接口和外部接口,前者是指软件模块之间的接口,后者是指供用户调用的接口,分体式医疗器械各独立部分的内部接口视为外部接口,如服务器与客户端、主机与从机的内部接口。从用户角度出发软件接口若无明示均指外部接口。
注册申请人需考虑软件接口的需求分析、风险管理、验证与确认、维护计划等活动要求,以及说明书与标签的设计要求。
需求分析基于软件接口的预期用户(如医务人员、患者、维护人员)、使用场景、预期用途明确其技术特征、使用限制。其中,软件接口包括供用户调用的应用程序接口、数据接口(含传输协议、存储格式,如DICOM、HL7、JPG、PNG、私有协议与格式)、产品接口(可联合使用的其他医疗器械独立软件、医疗器械硬件产品)。技术特征包括但不限于连接对象、信息内容、通信协议、性能指标、网络安全保证等要求。使用限制需考虑每个软件接口的预期用户范围、连接要求。
风险管理基于软件接口的预期用户、使用场景、预期用途及技术特征、使用限制予以实施,明确故障应对措施。验证与确认应保证软件接口满足设计要求,且已知剩余缺陷的风险均可接受。维护计划考虑软件接口的更新要求,软件接口更新亦需重新进行验证与确认。
说明书逐项说明每个软件接口的预期用户、使用场景、预期用途、技术特征、使用限制、故障应对措施。根据风险控制要求,标签明确关键软件接口的技术特征、使用限制。
注册申请人可单独提交一份互操作性研究资料,内容包括基本信息、需求规范、风险管理、验证与确认、维护计划;亦可在自研软件研究报告相关条款中体现软件接口要求,具体要求详见第八章。
(九)测量功能
测量功能(又称量化、定量功能)可分为图形学测量功能、客观物理测量功能,前者基于图形学间接反映客观事物的测量结果,后者直接反映客观事物的测量结果。无论何种测量功能均需结合测量的误差、不确定度等因素,明确测量准确性指标,如线性度、精度、重复性、再现性、范围限值、显示误差等。
医疗器械注册代理注册申请人需提供测量准确性的研究资料,并在说明书中向用户告知。此外,客观物理测量还需在产品技术要求中明确准确性指标,图形学测量还需在说明书中提供关于测量准确性的警示信息。
(十)远程访问与控制
对于远程访问与控制(含远程维护与升级)功能,需明确相关功能的性能指标要求和网络安全要求,具体要求详见医疗器械网络安全相关指导原则。
注册申请人需在软件研究资料、网络安全研究资料中提供相应研究资料,在产品技术要求中明确性能指标要求,并在说明书中向用户告知相应功能的使用要求(含网络安全)。
(十一)通用计算平台
通用计算平台本身不属于监管对象,需从风险管理角度考虑其对医疗器械的影响,具体要求取决于其是否作为医疗器械的产品结构组成。
对于独立软件、专用型独立软件视为软件组件,医疗器械的产品结构组成不含通用计算平台,在说明书中向用户告知通用计算平台需满足信息技术设备安全要求(含电磁兼容),并列明需符合的标准清单。
对于外控型软件组件,医疗器械的产品结构组成含有通用计算平台,在“其他研究资料”中提供通用计算平台满足信息技术设备安全要求(含电磁兼容)的证明性资料,如相关标准的自检报告、检测报告或相关认证文件。
(十二)非医疗器械功能
医疗器械软件若在技术上能够拆分非医疗器械功能,即采用模块化设计区分医疗器械功能和非医疗器械功能,则产品结构组成不应包含非医疗器械功能模块,说明书若含有非医疗器械功能应予以删除或注明为非医疗器械功能,产品技术要求不应含有非医疗器械功能。
医疗器械软件若在技术上无法拆分非医疗器械功能,需将非医疗器械功能作为自身组成部分予以整体考虑,重点关注非医疗器械功能对于医疗器械软件的影响及其风险。注册申请人需在医疗器械软件设计开发整体框架下考虑非医疗器械功能的软件生存周期过程质控要求,原则上可按软件安全性级别为轻微级别的要求予以处理。医疗器械软件的研究资料涵盖非医疗器械功能,说明书对非医疗器械功能予以注明,产品技术要求性能指标所述“功能”条款简述非医疗器械功能即可。
(十三)植入物产品设计软件
有些植入式医疗器械本身不含有医疗器械软件,但其安全有效性显著受限于产品设计软件的输出结果,如有源植入物设计软件(如可编程逻辑控件编程软件、集成电路设计软件)、个性化无源植入物(如骨科、齿科增材制造假体)设计软件等。
同时,有些植入式医疗器械需采用建模仿真软件进行安全有效性验证,如磁共振环境安全仿真软件、计算流体力学仿真软件、有限元计算仿真软件等。
因此,从医疗器械安全有效性评价角度出发,此两类植入物产品设计软件在植入式医疗器械注册申报时亦需提交软件研究资料。由于植入式医疗器械风险较高,植入物产品设计软件属于质量管理软件,故可参照严重级别的成品软件、自研软件要求提交软件研究资料,具体要求详见第八章。
(十四)使用期限
独立软件的使用期限即软件生存周期时限,通过商业因素予以确定,无需提供验证资料。
软件组件的使用期限与所属医疗器械相同,无需单独体现。专用型独立软件视为软件组件的使用期限要求与独立软件相同,在所属医疗器械使用期限研究资料中体现。
(十五)异常处理
异常处理(Exception handling)用于处理软件出现的异常状况,及时将软件异常信息告知用户,以采取风险控制措施保证安全有效性。注册申请人需在医疗器械软件设计开发过程中加强异常处理的设计工作,特别是在手术、急救等使用场景下。
(十六)功能安全与软件可靠性
考虑到行业实际情况,功能安全、软件可靠性暂不要求,待时机成熟时纳入考量。建议注册申请人参考相关标准要求加强功能安全、软件可靠性的设计工作,若已开展相应工作可在软件研究资料中予以提供。
(十七)GB/T 25000.51实施要求
GB/T 25000.51适用于医疗器械软件,其中“产品说明要求”、“用户文档集要求”适用于说明书,“软件质量要求”适用于软件本身,同时“使用质量”不适用。
注册申请人需在软件研究资料中提交GB/T 25000.51自测报告,亦可提交自检报告或检验报告代替自测报告,下同。
(十八)进口医疗器械软件
对于进口医疗器械软件,应提供本次申报软件在原产国(即注册地或生产地址所在国家/地区)获准上市的证明文件或证明性材料,注明软件完整版本。
进口医疗器械软件若存在中外差异,如语言差异、软件功能删减、适用范围缩减等,需在软件研究资料中提交本次申报软件与原产国获准上市版本软件的中外差异说明材料。
考虑到进口医疗器械软件不一定在中国同步注册,即该软件在原产国已多次变更注册但在中国为一次注册(含产品注册、变更注册)。对于产品注册,注册申报资料需涵盖本次申报软件的全部内容;对于变更注册,注册申报资料需涵盖软件自中国前次注册至本次申报的全部变更内容。
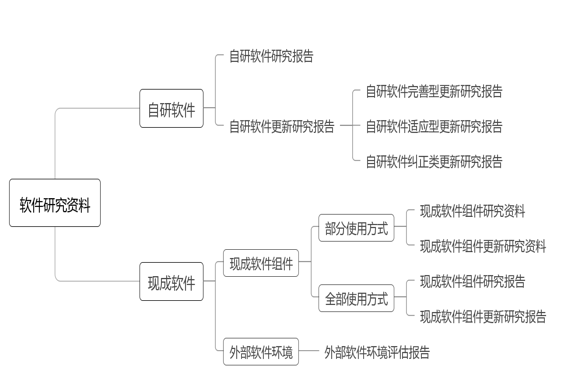
八、医疗器械软件研究资料
医疗器械软件研究资料框架详见图5,包括自研软件和现成软件(含现成软件组件、外部软件环境),具体要求如下。
(一)自研软件研究报告
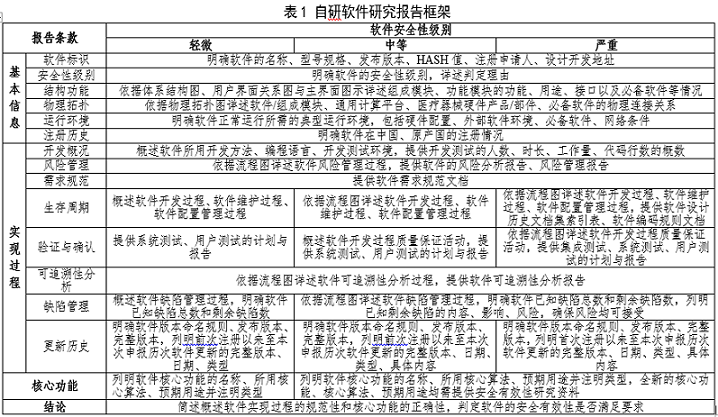
自研软件研究报告适用于自研软件的初次发布和再次发布,内容框架详见表1,包括基本信息、实现过程、核心功能、结论,详尽程度取决于软件安全性级别,不适用内容详述理由,附件均需注明软件完整版本。
[1]软件更新与软件版本命名规则若不匹配应采取纠正预防措施(CAPA)并予以记录。
[2]详见IMDRF/SaMD WG/N23 FINAL: 2015。
[3]平台功能软件即为特定功能软件的必备软件。
[4]详见IMDRF/SaMD WG/N41 FINAL: 2017。
[5]取代移动医疗器械指导原则关于云计算的要求。
图5 医疗器械软件研究资料框架
1. 基本信息
(1)软件标识
明确软件的名称、型号规格、发布版本、HASH值(如MD5值)以及注册申请人、设计开发地址。
(2)安全性级别
明确软件的安全性级别,详述判定理由。
(3)结构功能
基于软件设计规范文档提供软件的体系结构图、用户界面关系图与主界面图示,其中体系结构图区分医疗器械软件、必备软件、外部软件环境,用户界面关系图明确主界面、一级和二级用户界面的相互关系。
依据体系结构图详述图示软件模块(即组成模块)的功能、用途、接口以及必备软件、云计算等情况,并注明各组成模块的安全性级别。依据用户界面关系图(若适用)详述图示软件模块(即功能模块)的功能、用途、接口,依据主界面图示(若适用)详述主界面的布局、选项、功能。
若适用,组成模块和功能模块均需注明选装、模块版本。接口包括供用户调用的应用程序接口、数据接口、产品接口,逐项说明各接口的预期用户、使用场景、预期用途、技术特征、使用限制、故障应对措施。
(4)物理拓扑
基于软件设计规范文档提供软件的物理拓扑图(含云计算),依据物理拓扑图详述软件/组成模块、通用计算平台、医疗器械硬件产品/部件、必备软件之间的物理连接关系,包括全部外围设备。
(5)运行环境
明确软件(软件模块)正常运行所需的典型运行环境,包括硬件配置、外部软件环境、必备软件、网络条件。其中,硬件配置包括处理器、存储器、外设器件等要求;外部软件环境包括系统软件、通用应用软件、通用中间件、支持软件,注明全部软件的名称、完整版本、补丁版本,使用“兼容版本”而非“以上版本”、“更高版本”;若适用,必备软件明确名称、型号规格、发布版本、注册人;网络条件包括网络架构(如BS架构、CS架构、混合架构)、网络类型(如广域网、局域网、个域网)、网络带宽等要求。
若使用云计算,明确云计算的名称、服务模式、部署模式、配置以及云服务商的名称、住所、服务资质。
(6)注册历史
明确软件在中国、原产国的注册情况,列明历次注册的日期、发布版本、管理类别。软件组件明确所属医疗器械的注册情况。此外,亦可提供软件在其他主要国家和地区的注册情况。
2. 实现过程
(1)开发概况
概述软件所用开发方法(如面向过程、面向对象、敏捷开发等)、编程语言、开发测试环境(含软硬件设备、开发测试工具、网络条件、云计算),其中开发测试工具明确名称、完整版本、开发商;提供开发测试的人员总数、时长、工作量(人月数)、代码行总数的概数。
(2)风险管理
提供软件风险管理流程图,依据流程图详述软件风险管理过程的具体活动。提供软件的风险分析报告、风险管理报告,涵盖功能、性能、接口、运行环境、必备软件、云计算等情况,并提供采取风险控制措施前后的风险矩阵汇总表,另附软件开发所形成的原始文件。
软件组件提供所属医疗器械的风险管理文档,并注明软件组件所在位置。
(3)需求规范
提供软件需求规范文档,明确软件的功能、性能、接口、运行环境、必备软件、云计算等需求,另附软件开发所形成的原始文件。
软件组件若无单独文档,可提供所属医疗器械的产品需求规范文档,并注明软件组件所在位置。
(4)生存周期
轻微级别:概述软件开发过程、软件维护过程、软件配置管理过程的具体活动。
中等级别:提供软件开发、软件维护、软件配置管理流程图,依据流程图详述软件开发过程、软件维护过程、软件配置管理过程的具体活动。
严重级别:提供软件开发、软件维护、软件配置管理流程图,依据流程图详述软件开发过程、软件维护过程、软件配置管理过程的具体活动。提供软件设计历史文档集(DHF)索引表,另附软件编码规则文档。
此外,使用敏捷开发还需提供文件与记录控制文档。软件生存周期过程和活动亦可提供软件生存周期过程控制程序文档或软件生存周期过程标准核查表,用于替代相应描述。
(5)验证与确认
轻微级别:提供系统测试、用户测试的计划与报告。
中等级别:概述软件开发过程质量保证活动,并提供系统测试、用户测试的计划与报告。
严重级别:提供软件开发质量保证流程图,依据流程图详述软件开发过程的具体质量保证活动,并提供集成测试、系统测试、用户测试的计划与报告。
此外,测试计划和报告涵盖软件的功能、性能、接口、运行环境、必备软件、云计算等情况,另附软件开发所形成的原始文件。软件开发过程质量保证活动亦可提供软件开发质量保证计划文档,用于替代相应描述。
(6)可追溯性分析
提供软件可追溯性分析流程图,依据流程图详述软件可追溯性分析过程的具体活动。提供软件可追溯性分析报告,汇总列明软件需求规范文档、软件设计规范文档、源代码(明确软件单元名称即可)、软件测试报告、软件风险分析报告之间的对应关系,另附软件开发所形成的原始文件。
(7)缺陷管理
轻微级别:概述软件缺陷管理过程,明确软件已知缺陷总数和剩余缺陷数。
中等、严重级别:提供软件缺陷管理流程图,依据流程图详述软件缺陷管理过程的具体活动;明确软件已知缺陷总数和剩余缺陷数,列明软件已知剩余缺陷的内容、影响、风险,确保风险均可接受。软件已知剩余缺陷情况可另附文件。
(8)更新历史
轻微级别:提供软件版本命名规则,举例说明完整版本各字段含义,明确软件发布版本、软件完整版本;列明自前次注册以来至本次申报历次软件更新的完整版本、日期、类型。
中等级别:提供软件版本命名规则,举例说明完整版本各字段含义,明确软件发布版本、软件完整版本;列明自前次注册以来至本次申报历次软件更新的完整版本、日期、类型、具体内容。
严重级别:提供软件版本命名规则,举例说明完整版本各字段含义,明确软件发布版本、软件完整版本;列明自首次注册以来至本次申报历次软件更新的完整版本、日期、类型、具体内容。
此外,软件模块(含医用中间件)若单独进行版本控制,其版本命名规则亦需提供,并明确与软件版本命名规则的关系;软件和软件模块的版本命名规则均需与质量管理体系保持一致。软件更新类型注明重大更新或轻微更新。初次发布列明软件开发阶段历次软件更新情况。软件更新历史可另付文件。
3. 核心功能
轻微级别:基于说明书列明软件核心功能的名称、所用核心算法、预期用途,全新的核心功能、核心算法、预期用途均需注明。
中等、严重级别:基于说明书列明软件核心功能的名称、所用核心算法、预期用途,全新的核心功能、核心算法、预期用途均需注明,并提供相应安全有效性研究资料。其中,全新算法提供算法研究报告,通常包括算法基本信息、算法风险管理、算法需求规范、算法质控要求、算法验证与确认、算法可追溯性分析、结论等内容。测量功能提供测量准确性的研究资料。数据资源(如参考数据库)明确数据种类以及每类数据的样本量、数据分布等情况。
4. 结论
简述软件实现过程的规范性和核心功能的正确性,判定软件的安全有效性是否满足要求,受益是否大于风险。
(二)自研软件更新研究报告
自研软件更新研究报告适用于自研软件的再次发布,包括完善型更新、适应型更新、纠正类更新等研究报告。
完善型更新研究报告适用于自研软件发生重大、轻微完善型更新,或合并适应型更新、纠正类更新的情形,内容框架详见表2,不再赘述。
适应型更新研究报告适用于自研软件发生重大、轻微适应型更新,或合并纠正类更新,但未发生完善型更新的情形,内容包括软件标识、安全性级别、运行环境、风险管理、需求规范、生存周期、验证与确认、可追溯性分析、缺陷管理、更新历史、结论,具体要求详见表2相应说明。
纠正类更新研究报告适用于自研软件仅发生纠正类更新的情形,内容包括软件标识、安全性级别、风险管理、验证与确认、可追溯性分析、缺陷管理、更新历史、结论,具体要求详见表2相应说明。
考虑到软件更新具有累积效应,软件更新研究报告需涵盖医疗器械软件自前次注册(延续注册除外)以来软件更新的全部内容。
(三)现成软件研究资料
1.现成软件组件研究资料
(1)部分使用方式
对于部分使用方式,遗留软件、成品软件、外包软件的研究资料要求相同,无需单独提交研究报告,基于医疗器械软件的安全性级别,在自研软件研究报告适用条款中说明现成软件组件的情况。
适用条款包括软件标识、安全性级别、结构功能、运行环境、风险管理、需求规范、生存周期、验证与确认、可追溯性分析、缺陷管理、更新历史、核心功能、结论,其余条款若适用亦可予以说明。
此时若现成软件组件发生软件更新,完善型更新研究报告在自研软件完善型更新研究报告基础上,说明现成软件组件的变化情况,不适用条款说明理由;适应型更新、纠正类更新研究报告要求与自研软件相同。
(2)全部使用方式
对于全部使用方式,遗留软件、成品软件、外包软件的研究资料要求略有差异,单独提交现成软件组件研究报告及其类型判定依据。
现成软件组件研究报告条款与自研软件研究报告相同,但需基于现成软件组件(此时即医疗器械软件)的安全性级别予以说明,适用条款至少包括软件标识、安全性级别、运行环境、风险管理、需求规范、验证与确认、缺陷管理、核心功能、结论,不适用条款详述理由。遗留软件在验证与确认中还需提交其上市后使用问题(含不良事件、召回)的评价报告。
类型判定依据用于证明现成软件组件的类型。遗留软件提供《医疗器械生产质量管理规范》全面实施日期(2018年1月1日)之前的产品注册证信息或产品上市证明性材料。成品软件提供外购合同等证明性材料,若已在中国上市提供产品注册证信息。外包软件提供外包合同或协议等证明性材料。
此时若现成软件组件发生软件更新,完善型更新研究报告在现成软件组件研究报告基础上,说明现成软件组件的变化情况,不适用条款说明理由;适应型更新、纠正类更新研究报告要求与自研软件相同。
2.外部软件环境评估报告
外部软件环境评估报告用于医疗器械软件外部软件环境(含云计算)的评估,适用于医疗器械软件的初次发布和再次发布,内容框架详见表3,包括安全性级别、软件标识、功能用途、运行环境、风险管理、验收管理、维护计划、结论,详尽程度取决于软件安全性级别。
(1)安全性级别
依据医疗器械软件安全性级别,明确外部软件环境的安全性级别。
(2)软件标识
按照系统软件、应用软件、中间件、支持软件四种类型,分类描述外部软件环境所含全部现成软件的名称、完整版本、补丁版本、发布日期、供应商。
(3)功能用途
按照系统软件、应用软件、中间件、支持软件四种类型,分类描述外部软件环境所含全部现成软件的功能、用途、与医疗器械软件的关系、使用限制、选择依据。
(4)运行环境
按照系统软件、应用软件、中间件、支持软件四种类型,分类描述外部软件环境所含全部现成软件的运行环境,结合兼容性考虑医疗器械软件运行环境的确定依据。
(5)风险管理
提供外部软件环境所含全部现成软件的风险分析报告,另附软件开发所形成的原始文件。可提供医疗器械软件的风险分析报告,并注明外部软件环境所在位置。
(6)验收管理
轻微级别:概述外部软件环境验收管理过程相关活动。
中等级别:依据流程图详述外部软件环境验收管理过程相关活动。
严重级别:依据流程图详述外部软件环境验收管理过程相关活动,提供外部软件环境兼容性测试计划和报告,另附软件开发所形成的原始文件。
(7)维护计划
轻微级别:概述外部软件环境更新管理过程相关活动,包括补丁更新、版本更新、产品更新。
中等级别:依据流程图详述外部软件环境更新管理过程相关活动,包括补丁更新、版本更新、产品更新。
严重级别:依据流程图详述外部软件环境更新管理过程相关活动,包括补丁更新、版本更新、产品更新;提供现成软件停运后续维护方案,即现成软件供应商停止售后服务后,注册申请人对于现成软件的维护方案,如云计算服务终止后的无损数据迁移方案。
(8)结论
简述外部软件环境所含全部现成软件的质量是否满足要求。
表3 外部软件环境评估报告框架
|
报告条款 |
软件安全性级别 |
|
轻微 |
中等 |
严重 |
|
安全性级别 |
依据医疗器械软件安全性级别,明确外部软件环境的安全性级别 |
|
软件标识 |
分类描述各现成软件的名称、完整版本、补丁版本、发布日期、供应商 |
|
功能用途 |
分类描述各现成软件的功能、用途、与医疗器械软件的关系、使用限制、选择依据 |
|
运行环境 |
分类描述各现成软件的运行环境,明确医疗器械软件运行环境的确定依据 |
|
风险管理 |
提供各现成软件的风险分析报告 |
|
验收管理 |
概述外部软件环境验收管理过程 |
依据流程图详述外部软件环境验收管理过程 |
依据流程图详述外部软件环境验收管理过程,提供兼容性测试计划和报告 |
|
维护计划 |
概述外部软件环境更新管理过程 |
依据流程图详述外部软件环境更新管理过程 |
依据流程图详述外部软件环境更新管理过程,提供现成软件停运后续维护方案 |
|
结论 |
简述外部软件环境所含全部现成软件的质量是否满足要求 |
外部软件环境发生更新亦需依据其安全性级别开展相应评估工作,并更新医疗器械软件的外部软件环境评估报告,以备后续体系核查或变更注册使用。
九、注册申报资料补充说明
医疗器械注册办理申报资料在符合医疗器械注册申报资料要求等文件要求基础上,重点关注以下要求。
(一)产品注册
1. 申请表信息
(1)独立软件
产品名称应符合独立软件通用名称命名规范要求,通常体现输入数据、核心功能、预期用途等特征词。
型号规格注明软件发布版本,无需体现版本英文缩写V。
结构组成明确交付内容和功能模块,其中交付内容包括软件安装程序、授权文件、外部软件环境安装程序等软件程序文件,功能模块包括客户端、服务器端(若适用)、云端(若适用),若适用注明选装、模块版本。
适用范围通常基于预期用途、使用场景、核心功能予以规范,若适用分述各功能模块的适用范围。同时保证用语的规范性,区分“分析”与“测量”、“手术模拟”与“手术计划”,使用“显示”、“接收”而非“浏览”、“采集”。
(2)软件组件
软件组件通常无需在注册证载明信息中体现。其软件功能名称可参照独立软件要求。结构组成保证用语规范性,使用“主机”、“工作站”而非“电脑”、“计算机”。若有辅助决策类软件功能,结构组成(若适用)和适用范围需予以体现。
对于专用型独立软件视为软件组件的情形,软件名称与独立软件要求相同,结构组成明确软件的名称、型号规格、发布版本,适用范围体现辅助决策类软件功能。
2. 软件研究资料
提交自研软件研究报告、外部软件环境评估报告(若适用)以及GB/T 25000.51自测报告。
若使用现成软件组件,根据其使用方式提交相应研究资料。相关研究资料的具体要求详见第八章。
此外,鼓励在软件研究资料中提交医疗器械产品市场宣传材料。该材料仅作为审评参考材料以补充产品信息,不作为审评对象,亦不作为审评决策依据。
3. 产品技术要求
(1)独立软件
独立软件产品技术要求 “产品型号/规格及其划分说明”明确软件的名称、型号规格、发布版本、版本命名规则,软件模块(含医用中间件)若有单独的版本、版本命名规则均需说明。
“性能指标”包括通用要求、专用要求、安全要求,其中通用要求根据软件产品特性进行规范,不适用内容在非临床资料产品技术要求章节中予以说明;专用要求符合相关产品标准(如放射治疗计划软件)要求,安全要求符合相关安全标准(如报警、放射治疗)要求。
“附录”提供体系结构图、用户界面关系图与主界面图示、物理拓扑图以及必要的注释。
独立软件产品技术要求模板详见附录。
(2)软件组件
软件组件在所属医疗器械的产品技术要求中进行规范,其中“产品型号/规格及其划分说明”明确软件的名称、型号规格(若适用)、发布版本、版本命名规则,软件模块(含医用中间件)若有单独的版本、版本命名规则均需说明。
“性能指标”包括软件的功能、使用限制、接口、访问控制、运行环境(若适用)、性能效率(若适用)等要求。其中,功能明确软件的全部核心功能(含安全功能)纲要,注明选装、自动功能,其中客观物理测量功能明确测量准确性指标,数据资源(如参考数据库)明确数据种类和每类数据的样本量,若核心功能相同但核心算法类型不同则每类核心算法均需备注;使用限制包括用户使用限制和技术限制;接口包括供用户调用的应用程序接口、数据接口、产品接口;访问控制明确软件的用户身份鉴别方法、用户类型及用户访问权限;运行环境、性能效率适用于外控型软件组件、专用型独立软件视为软件组件,其中运行环境(含云计算)明确典型配置,包括硬件配置、外部软件环境、网络条件,性能效率明确软件在典型运行环境下完成典型核心功能的时间特性,若适用明确资源利用性、容量。上述条款不适用内容在非临床资料予以说明。
对于专用型独立软件视为软件组件,除上述软件组件要求外,还需在“附录”中提供体系结构图、用户界面关系图与主界面图示、物理拓扑图以及必要的注释。
4. 说明书
说明书应符合相关法规、规范性文件、国家标准、行业标准要求。
说明书需体现软件的功能、使用限制、输入输出数据类型、必备软硬件、最大并发数、接口、访问控制、运行环境(若适用)、性能效率(若适用)等信息,明确软件发布版本。
其中,软件功能包括全部核心功能(含安全功能),注明选装、自动功能,其中测量功能明确测量准确性指标,图形学测量功能还需提供关于测量准确性的警示信息,数据资源明确数据种类和每类数据的样本量。接口逐项说明每个供用户调用软件接口的预期用户、使用场景、预期用途、技术特征、使用限制、故障应对措施。运行环境(含云计算)、性能效率适用于独立软件、外控型软件组件、专用型独立软件视为软件组件,具体要求详见前文。
若适用,向用户告知通用计算平台满足信息技术设备安全要求(含电磁兼容),并列明相应标准清单。
5. 产品标签样稿(独立软件适用)
对于物理交付方式,产品标签应符合相应规定。对于网络交付方式,提交产品网络交付页面照片,该页面的产品注册信息应符合相应规定。
此外,建议在“关于”或“帮助”等软件用户界面体现产品注册信息。
(二)变更注册
1. 软件研究资料
医疗器械变更注册应根据软件更新情况,提交软件变化部分对产品安全性与有效性影响的研究资料:
(1)涉及完善型软件更新:适用于自研软件发生完善型更新,或合并适应型更新、纠正类更新的情形,此时提交自研软件完善型更新研究报告(或自研软件研究报告)、外部软件环境评估报告(若适用)以及GB/T 25000.51自测报告;
(2)涉及适应型软件更新:适用于自研软件发生适应型更新,或合并纠正类更新,但未发生完善型更新的情形,此时提交自研软件适应型更新研究报告(或自研软件研究报告);
(3)仅发生纠正类软件更新:适用于自研软件仅发生纠正类更新的情形,此时提交自研软件纠正类更新研究报告;
(4)未发生软件更新:出具真实性声明,明确对此承担法律责任。
若使用现成软件组件,根据其使用方式提交相应研究资料。相关研究资料的具体要求详见第八章。
若适用,鼓励在软件研究资料中提交医疗器械产品市场宣传材料。该材料仅作为审评参考材料以补充产品信息,不作为审评对象,亦不作为审评决策依据。
2. 产品技术要求
(1)独立软件
独立软件产品技术要求体现软件更新情况,包括“产品型号/规格及其划分说明”、“性能指标”、“附录”。
(2)软件组件
软件组件在所属医疗器械的产品技术要求中体现软件更新情况,包括“产品型号/规格及其划分说明”的软件信息、“性能指标”的软件要求。
专用型独立软件视为软件组件的要求与软件组件相同。
3. 说明书
若适用,提交说明书变化情况说明。
4. 产品标签样稿(独立软件适用)
若适用,提交申报产品的产品标签样稿及其变化情况说明。
(三)延续注册
延续注册通常无需提交软件相关研究资料。若适用,根据注册证“备注”所载明的要求提交相应软件研究资料。
产品技术要求“产品型号/规格及其划分说明”明确软件的名称、型号规格、发布版本、版本命名规则,若原注册产品标准(或原产品技术要求)及其变更对比表未体现上述软件信息,需在符合性声明中予以明确。
独立软件产品技术要求“性能指标”、“检验方法”无需按照本指导原则附录要求进行修改,应与原注册产品标准(或原产品技术要求)及其变更对比表保持一致,且“性能指标”相关条款删除注册证已载明信息。
十、参考文献
[1]原国家食品药品监督管理总局.医疗器械说明书和标签管理规定(总局令第6号)[Z],2014.7
[2]原国家食品药品监督管理总局.医疗器械分类规则(总局令第15号)[Z],2015.7
[3]原国家食品药品监督管理总局.医疗器械召回管理办法(总局令第29号)[Z],2017.1
[4]国家市场监督管理总局. 医疗器械不良事件监测和再评价管理办法(总局令第1号)[Z],2018.8
[5]国家市场监督管理总局.医疗器械注册与备案管理办法(总局令第47号)[Z],2021.8
[6]原国家食品药品监督管理总局.医疗器械生产质量管理规范(2014年第64号公告)[Z],2014.12
[7]国家药品监督管理局.医疗器械唯一标识系统规则(2019年第66号公告)[Z],2019.8
[8]国家市场监督管理总局.医疗器械注册申报资料要求和批准证明文件格式(2021年第121号公告)[Z],2021.9
[9] 国家药品监督管理局.医疗器械注册自检管理规定(2021年第126号公告)[Z],2021.10
[10]原国家食品药品监督管理总局.医疗器械软件注册技术审查指导原则(2015年第50号通告)[Z],2015.8
[11]原国家食品药品监督管理总局.医学图像存储传输软件(PACS)注册技术审查指导原则(2016年第27号通告)[Z],2016.3
[12]原国家食品药品监督管理总局.医疗器械网络安全注册技术审查指导原则(2017年第13号通告)[Z],2017.1
[13]原国家食品药品监督管理总局.医疗器械注册单元划分指导原则(2017年第187号通告)[Z],2017.11
[14]原国家食品药品监督管理总局.中央监护软件注册技术审查指导原则(2017年第198号通告)[Z],2017.12
[15]原国家食品药品监督管理总局.移动医疗器械注册技术审查指导原则(2017年第222号通告)[Z],2017.12
[16]国家药品监督管理局.有源医疗器械使用期限注册技术审查指导原则(2019年第23号通告)[Z],2019.5
[17]国家药品监督管理局.医疗器械生产质量管理规范附录独立软件(2019年第43号通告)[Z],2019.7
[18]国家药品监督管理局. 医疗器械通用名称命名指导原则(2019年第99号通告)[Z],2019.12
[19]国家药品监督管理局.医疗器械安全和性能的基本原则(2020年第18号通告)[Z],2020.3
[20]国家药品监督管理局. 医用软件通用名称命名指导原则(2021年第48号通告)[Z],2021.7
[21]国家药品监督管理局.医疗器械临床评价技术指导原则(2021年第73号通告)[Z],2021.9
[22]国家药品监督管理局. 医疗器械产品技术要求编写指导原则(2022年第8号通告)[Z],2022.2
[23]国家药品监督管理局.医疗器械生产质量管理规范独立软件现场检查指导原则(药监综械管〔2020〕57号)[Z],2020.5
[24]国家药品监督管理局医疗器械技术审评中心. 医疗器械人因设计技术审查指导原则(征求意见稿)[Z],2020.5
[25]国家药品监督管理局医疗器械技术审评中心. 深度学习辅助决策医疗器械软件审评要点(2019年第7号通告)[Z],2019.7
[26]国家药品监督管理局医疗器械技术审评中心. 肺炎CT影像辅助分诊与评估软件审评要点(试行)(2020年第8号通告)[Z],2020.3
[27] GB 4793.1-2007测量、控制和实验室用电气设备的安全要求 第1部分:通用要求[S]
[28] GB 4943.1-2011信息技术设备 安全 第1部分:通用要求[S]
[29] GB9706.1-2020 医用电气设备 第1部分:基本安全和基本性能的通用要求[S]
[30]GB 16174.1-2015手术植入物有源植入式医疗器械 第1部分:安全、标记和制造商所提供信息的通用要求[S]
[31] GB/T 8566-2007信息技术 软件生存周期过程[S]
[32] GB/T 9254-2008信息技术设备的无线电骚扰限值和测量方法[S]
[33] GB/T 11457-2006信息技术 软件工程术语[S]
[34] GB/T 18268.1-2010测量、控制和实验室用的电设备 电磁兼容性要求 第1部分:通用要求[S]
[35] GB/T 19003-2008软件工程 GB/T 19001-2000应用于计算机软件的指南[S]
[36] GB/T 20157-2006信息技术 软件维护[S]
[37] GB/T 20158-2006信息技术 软件生存周期过程 配置管理[S]
[38] GB/T 20438.1-2017电气/电子/可编程电子安全相关系统的功能安全 第1部分:一般要求[S]
[39]GB/T 20438.2-2017电气/电子/可编程电子安全相关系统的功能安全 第2部分:电气/电子/可编程电子安全相关系统的要求[S]
[40] GB/T20438.3-2017电气/电子/可编程电子安全相关系统的功能安全 第3部分:软件要求[S]
[41] GB/T20438.4-2017电气/电子/可编程电子安全相关系统的功能安全 第4部分:定义和缩略语[S]
[42] GB/T 20918-2007信息技术 软件生存周期过程 风险管理[S]
[43] GB/T 25000.10-2016系统与软件工程 系统与软件质量要求和评价(SQuaRE) 第10部分:系统与软件质量模型[S]
[44] GB/T 25000.12-2017 系统与软件工程 系统与软件质量要求和评价(SQuaRE)第12部分:数据质量模型[S]
[45] GB/T 25000.51-2016系统与软件工程 系统与软件质量要求和评价(SQuaRE) 第51部分:就绪可用软件产品(RUSP)的质量要求和测试细则[S]
[46]GB/T 29832.1-2013 系统与软件可靠性 第1部分:指标体系[S]
[47]GB/T 29832.2-2013 系统与软件可靠性 第2部分:度量方法[S]
[48]GB/T 29832.3-2013 系统与软件可靠性 第3部分:测试方法[S]
[49] GB/T 31167-2014信息安全技术 云计算服务安全指南[S]
[50] GB/T 31168-2014信息安全技术 云计算服务安全能力要求[S]
[51]GB/T 32422-2015 软件工程 软件异常分类指南[S]
[52] GB/T 35278-2017信息安全技术 移动终端安全保护技术要求[S]
[53]YY 0505-2012医用电气设备 第1-2部分:安全通用要求 并列标准:电磁兼容 要求和试验[S]
[54] YY 0637-2013医用电气设备 放射治疗计划系统的安全要求[S]
[55] YY 0709-2009医用电气设备 第1-8部分:安全通用要求 并列标准:医用电气设备和医用电气系统中报警系统的测试和指南[S]
[56] YY 0721-2009医用电气设备 放射治疗记录与验证系统的安全[S]
[57] YY 0775-2010远距离放射治疗计划系统 高能X(γ)射束剂量计算准确性要求和试验方法[S]
[58] YY/T 0287-2017医疗器械 质量管理体系 用于法规的要求[S]
[59] YY/T 0316-2016医疗器械 风险管理对医疗器械的应用[S]
[60] YY/T 0664-2020 医疗器械软件 软件生存周期过程[S]
[61] YY/T 0887-2013放射性粒籽植入治疗计划系统剂量计算要求和试验方法[S]
[62] YY/T 0889-2013调强放射治疗计划系统性能和试验方法[S]
[63] YY/T 0973-2016自动控制式近距离后装设备放射治疗计划系统 性能和试验方法[S]
[64] YY/T 1406.1-2016医疗器械软件 第1部分:YY/T 0316应用于医疗器械软件的指南[S]
[65] YY/T 1630-2018医疗器械唯一标识基本要求[S]
[66] YY/T 1681-2019医疗器械唯一标识系统基础术语[S]
[67] IMDRF/UDI WG/N7 FINAL: 2013, UDI Guidance: Unique Device Identification of Medical Devices[Z], 2013.9
[68] IMDRF/SaMD WG/N10 FINAL: 2013, SaMD: Key Definitions[Z], 2013.12
[69] IMDRF/SaMD WG/N12 FINAL: 2014, SaMD: Possible Framework for Risk Categorization and Corresponding Considerations[Z], 2014.9
[70] IMDRF/SaMD WG/N23 FINAL: 2015, SaMD: Application of Quality Management System[Z], 2015.10
[71] IMDRF/SaMD WG/N41 FINAL: 2017, SaMD: Clinical Evaluation[Z], 2017.9
[72] IMDRF/CYBER WG/N60FINAL: 2020, Principles and Practices for Medical Device Cybersecurity[Z], 2020.4
[73] FDA, General Principles of Software Validation[Z], 2002.1
[74] FDA, Cybersecurity for Networked Medical Devices Containing Off-the-Shelf Software[Z], 2005.1
[75] FDA, Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices[Z], 2005.5
[76] FDA, Modifications to Devices Subject to Premarket Approval(PMA)[Z], 2008.12
[77] FDA, Acceptable Media for Electronic Product User Manuals[Z], 2010.3
[78] FDA, Computer-Assisted Detection Devices Applied to Radiology Images and Radiology Device Data[Z], 2012.7
[79] FDA, Considerations for Computer-Assisted Detection Devices Applied to Radiology Images and Radiology Device Data[Z], 2012.7
[80] FDA, Content of Premarket Submissions for Management of Cybersecurity in Medical Devices[Z], 2014.10
[81] FDA, Postmarket Management of Cybersecurity in Medical Devices[Z], 2016.12
[82] FDA, Applying Human Factors and Usability Engineering to Medical Devices[Z], 2016.2
[83] FDA, Deciding When to Submit a 510(k) for a Software Change to an Existing Device[Z], 2017.10
[84] FDA, Design Considerations and Pre-market Submission Recommendations for Interoperable Medical Devices[Z], 2017.9
[85] FDA, Medical Device Accessories - Describing Accessories and Classification Pathways[Z], 2017.12
[86] FDA, Technical Considerations for Additive Manufactured Medical Devices[Z], 2017.12
[87] FDA, Content of Premarket Submissions for Management of Cybersecurity in Medical Devices(Draft Guidance)[Z], 2018.10
[88] FDA, Developing a Software Precertification Program: A Working Model[Z], 2019.1
[89] FDA, Technical Performance Assessment of Quantitative Imaging in Device Premarket Submission(Draft Guidance)[Z], 2019.4
[90] FDA, Off-The-Shelf Software Use in Medical Devices[Z], 2019.9
[91] FDA, General Wellness: Policy for Low Risk Devices[Z], 2019.9
[92] FDA, Medical Device Data Systems, Medical Image Storage Devices, and Medical Image Communications Devices[Z], 2019.9
[93] FDA, Policy for Device Software Functions and Mobile Medical Applications[Z], 2019.9
[94] FDA, Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act[Z], 2019.9
[95] FDA, Clinical Decision Support Software(Draft Guidance)[Z], 2019.9
[96] FDA, Multiple Function Device Products: Policy and Considerations[Z], 2020.7
[97] FDA, Artificial Intelligence and Machine Learning (AI/ML)Software as a Medical Device(SaMD) Action Plan[Z], 2021.1
[98] FDA, Content of Premarket Submissions for Device Software Functions(Draft Guidance)[Z], 2021.11
[99] FDA. Technical Considerations for Medical Devices with Physiologic Closed-Loop Control Technology(Draft Guidance)[Z], 2021.12
[100] FDA. Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions(Draft Guidance)[Z], 2021.12
[101] FDA, FDASIA Health IT Report: Proposed Strategy and Recommendations for a Risk-Based Framework[Z], 2014.4
[102] FDA, Report on Non-Device Software Functions: Impact to Health and Best Practices[Z], 2020.11
[103] MEDDEV 2.1/6, Qualification and Classification of Standalone Software[Z], 2016.7
[104] MDCG 2019-11, Guidance on Qualification and Classification of Software in Regulation(EU) 2017/745 - MDR and Regulation(EU) 2017/746 - IVDR[Z], 2019.10
[105]MDCG 2020-1, Guidance on clinical evaluation(MDR)/ Performance evaluation(IVDR) of medical device software[Z], 2020.3
[106] AAMI TIR45:2012/(R)2018, Guidance on the use of AGILE practices in the development of medical device software[S]
[107] ISO TR 17791:2013, Health informatics - Guidance on standards for enabling safety in health software[S]
[108] IEC 62304 AMD1:2015, Medical device software - Software life cycle processes[S]
[109] IEC 62366-1:2015 Medical devices - Part 1: Application of usability engineering to medical devices[S]
[110] IEC 80001-1:2010, Application of risk management for IT-networks incorporating medical devices - Part 1: Roles, responsibilities and activities[S]
[111] ISOTR 80002-2:2017, Medical device software - Part 2: Validation of software for regulated processes[S]
[112] IECTR 80002-3:2014, Medical device software - Part 3: Process reference model of medical device software life cycle processes(IEC 62304)[S]
[113] IEC 82304-1:2016, Health Software - Part 1: General requirements for product safety[S]
[114] ISO TS 82304-2:2021, Health software - Part 2: Health and wellness apps - Quality and reliability[S]
附录:独立软件产品技术要求模板
医疗器械产品技术要求
医疗器械产品技术要求编号:
产品名称
1. 产品型号/规格及其划分说明
1.1 软件型号规格
明确软件的型号/规格,无需体现软件发布版本。
1.2 软件发布版本
明确软件发布版本,若软件模块(含医用中间件)单独进行版本控制亦需提供其发布版本。
1.3 软件版本命名规则
明确软件完整版本全部字段的位数、范围、含义,若软件模块(含医用中间件)单独进行版本控制亦需提供其版本命名规则,并明确与软件版本命名规则的关系。软件和软件模块的版本命名规则均需与质量管理体系保持一致。
2. 性能指标
2.1 通用要求
2.1.1 功能
依据说明书和用户界面明确软件供用户调用的全部功能(含安全功能)纲要,注明选装、自动功能,其中客观物理测量功能明确测量准确性指标,数据资源(如参考数据库)明确数据种类和每类数据的样本量。若核心功能相同但核心算法类型不同,则每类核心算法均需备注。
2.1.2 使用限制
依据说明书明确软件的用户使用限制和技术限制。
2.1.3 输入输出
明确软件的输入数据类型(如医学图像、生理参数、体外诊断等数据)、输出结果类型(如处理、测量、分析等结果)。
2.1.4 接口
明确软件供用户调用的应用程序接口(API)、数据接口(含传输协议、存储格式,如DICOM、HL7、JPG、PNG、私有协议与格式)、产品接口(可联合使用的其他医疗器械独立软件、医疗器械硬件产品)。
2.1.5 必备软硬件
明确软件正常运行所必需的其他的医疗器械独立软件(名称、型号规格、发布版本)及医用中间件(名称、型号规格、发布版本)、医疗器械硬件产品(名称、型号规格)。
2.1.6 运行环境
明确软件正常运行所需的典型运行环境,包括硬件配置(含处理器、存储器、外设器件)、外部软件环境(列明全部软件的名称、完整版本、补丁版本,使用“兼容版本”而非“以上版本”、“更高版本”)、网络条件(含网络架构、网络类型、网络带宽),涵盖客户端、服务器端(若适用)、云端(若适用)要求。无需重复描述必备软硬件。
2.1.7 性能效率
明确软件在典型运行环境(含云计算)下完成典型核心功能的时间特性,若适用明确资源利用性、容量。
2.1.8 最大并发数
明确软件在典型运行环境(含云计算)下的实施典型并发操作的最大并发用户数和/或患者数,注明相应响应时间。
2.1.9 用户界面
明确软件的用户界面类型和用户输入类型。
2.1.10 消息
明确软件向用户提供的消息类型和形式。
2.1.11用户差错防御
明确软件对导致严重后果的用户操作错误的防御能力。
2.1.12 访问控制
明确软件的用户身份鉴别方法、用户类型及用户访问权限。
2.1.13 版权保护
明确软件的版权保护技术及其对软件正常使用的影响。
2.1.14 可靠性
明确软件出错的数据保存、恢复及继续运行能力。
2.1.15 维护性
明确软件向用户提供的维护功能和维护信息类型。
2.2 专用要求 (若适用)
注:依据专用标准(名称、发布年份)适用条款逐条描述
2.2.1 YY 0775-2010(若适用)
2.2.2 YY/T 0887-2013(若适用)
2.2.3 YY/T 0889-2013(若适用)
2.2.4 YY/T 0973-2016(若适用)
……
2.3 安全要求 (若适用)
注:明确安全标准名称和发布年份
2.3.1 YY 0709-2009(若适用)
2.3.2 YY 0637-2013(若适用)
2.3.3 YY 0721-2009(若适用)
……
3. 检验方法
3.0 依据检测单元分述软件测试环境(与典型运行环境等同)。
3.1 通用要求符合性检验
通过检查说明书、实际操作、软件测试等方法逐条说明2.1各条款的检验方法,并验证2.1各条款的符合性。
若核心功能相同但核心算法类型不同,则每类核心算法所对应的核心功能均需检测(检测对象为核心功能而非核心算法)。
3.2 专用要求检验方法(若适用)
3.2.1依据YY 0775-2010的方法进行检验(若适用)。
3.2.2依据YY/T 0887-2013的方法进行检验(若适用)。
3.2.3依据YY/T 0889-2013的方法进行检验(若适用)。
3.2.4依据YY/T 0973-2016的方法进行检验(若适用)。
……
3.3 安全要求检验方法(若适用)
3.3.1 依据YY 0709-2009的方法进行检验(若适用)。
3.3.2 依据YY 0637-2013的方法进行检验(若适用)。
3.3.3 依据YY 0721-2009的方法进行检验(若适用)。
……
4. 术语(若适用)
注:明确软件所用专业术语(缩写)含义
4.1 ……
4.2 ……
4.3 ……
……
(分页)
附录
注:图示均需提供必要注释
1.体系结构图
2.用户界面关系图与主界面图示
3.物理拓扑图
鸿远医疗器械咨询辅导成功案例
深圳鸿远医疗器械咨询有限公司是一家技术专业的 医疗器械咨询公司,专注提供全国各地如:深圳、广州、东莞、中山、佛山、潮州、顺德、广西、上海、西安、重庆、成都等知名城市的医疗器械领域技术咨询服务。鸿远医疗器械咨询专业服务于: 医疗器械产品注册证、医疗器械生产许可证、进口医疗器械注册、一类医疗器械产品备案及生产备案代办、医疗器械经营许可证代办、二类医疗器械经营备案、医疗器械分类界定、CE认证、ISO13485认证、FDA注册、临床试验、医疗器械质量管理体系认证及体系建立与过程确认文件建立(ISO9001, ISO13485, GMP, CE,QSR820,CMDCAS);产品技术要求制订、技术文件、临床试验及免临床同类产品比对比资料编写、注册资料编写辅导、电磁兼容预测整改、医疗器械广告批文申报、医疗器械出口销售证明办理、洁净室建设指导等提供一站式服务,技术专业,诚信服务,欢迎您咨询!
|
|
|
|
|
|