|
现代化临床试验的实施与监管
一、引言
在过去的 20 年里,临床试验的数量和复杂程度呈现出极速增长的趋势,各大企业、研究中心及监管机构亦在积极地探索更加高效科学的方法来管理、实施以及审评临床试验。近年来,随着我国创新药品,生物制剂及医疗器械产品快速发展,在能有效保障受试者权益,获得科学真实临床数据的基础上,完成一项高质量的临床试验成为了一项重大挑战。2017 年发布的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》中也对监管部门提出要求,探索临床试验监管方法的改革道路,加快临床试验进程。
2013 年 8 月,美国食品药品监督管理局(Food and Drug Administration, FDA)正式颁布了新的临床试验监察指导原则 -A Risk-Based Approach to Monitoring,第一次从监管的角度向行业推荐基于风险的临床试验监察(Risk-Based Monitoring, RBM) 的 理念,代表着 FDA 对临床试验管理方法的有效指引和大胆尝试。
一个新产品在研发过程中相当大一部分成本需要投入在临床试验中,以往研究发现,临床试验总费用的 25%-30% 消耗在现场监察(on-site monitoring)中 [1],而其对提升临床试验质量的意义却存在争议,RBM原则鼓励申办者在合适的条件下更多地使用中心化监察(centralized monitoring),即通过对纸质版病例报告表(CRF)或电子数据采集系统(eDC)收集到的数据进行趋势分析,得到临床试验的风险点和关键性数据点,指导申办方分配资源,降低成本,高效完成研究。
二、目前我国临床研究的困境
目前,我国临床研究主要面临以下困境:首先,我国临床资源分布不均导致研究者对临床试验要求的理解程度以及 GCP 意识的强弱均存在很大的差异[2];其次,eDC 系统的使用未得到普及;再次,申办者质控能力以及临床监察员素质不均衡。基于以上因素,大部分国内的申办者选择100% 的对临床试验原始数据进行核查,这种做法成本极高且效率低下,因为部分非关键性数据,如受试者基线身高、体重等并不会对试验结果产生重要影响,也不会影响审评机构对结果的判断。
而国际上很多先进企业已经选择 RBM 模式开展临床试验监察,在保障数据完整、真实的基础上用更少的人力成本完成试验,并且得到了监管机构的认可。如果我国临床试验不及时跟上现代化的脚步,在未来很有可能会遇到发展上的障碍。
三、基于风险监察模式的优势及可行性
基于风险监察的优势主要有一下几点:
第一,实时收集数据,更早发现错误,保障受试者安全及方案依从性。传统监察方法一般要求申办者每 4-8 周进行一次现场监察,来发现并纠正此期间发生的方案偏离,这种方法存在严重的滞后性[3];
第二,节省资源,更高效的识别问题。一项回顾性研究证明中心化监察可以发现90% 现场监察能发现的问题,另外,2012 年,FDA 和 EMA(European Medicines Agency)开展的一项研究发现[4],中心化监察可以保证几乎100% 的方案依从,方案设计上的不足在入组少量受试者时即可被发现并纠正,eDC 的自动校正和逻辑检查功能在早期即可发现并减少数据错误、缺失的发生;
第三,建立数据库,大量运用 eDC 系统可以收集宝贵的临床使用数据,助力日后更加系统的研究及产品开发,甚至开发人工智能系统帮助监管及审评决策。
相比较新药研发的临床试验,医疗器械的临床试验的一些特点使RBM 模式更具有可行性。首先医疗器械临床试验的数据点远远少于药品临床试验,关键性的数据主要包括主要研究终点、严重不良事件以及非预期的不良事件,风险分析相对简单;其次,医疗器械的临床试验部分研究终点以影像学的形式确定,如 MRI,DSA,CT 等等,这些量化的数据更方便软件处理以及共享上传;最后,近年来我国互联网及软件产品迅猛发展,eDC 系统的使用开始增多,与医院电子病历系统的兼容性变强。以上因素均可以保障基于风险的临床试验监察监管得以实现。
四、基于风险监察模式的应用方法
不论是申办者组织的日常监察还是医疗器械监管机构开展的临床试验现场核查,关注的重点基本一致,主要包括:病例报告表数据与原始数据一致性,数据缺失,数据造假,受试者知情同意合规性,研究器械管理合规性,不良事件记录及上报合规性,以及数据与申报资料的一致性。下面将举例说明 RBM 的应用方法。
1. 筛选高风险研究中心
如果发现某一家研究中心入组速度远超过其他中心,可能的风险点则可能是受试者是否完全满足入排标准,研究者有没有按照 ICH-GCP 的要求给受试者知情同意,此时系统会触发风险警报,提示该中心需要尽快安排现场监察或与研究者加强电话沟通。
2. 识别高风险数据错误及其趋势
申办者根据研究方案、试验器械风险级别、主要研究终点确定高风险数据点,在系统中设置常规值,如果研究者输入的实测值超出范围会出现提示,迫使研究者进行及时再次确认,如果某一数据在各个时间点没有任何差异,则提示数据造假可能,如果差异过大,则提示检测方法错误或数据造假可能。
3. 早期提示不良事件
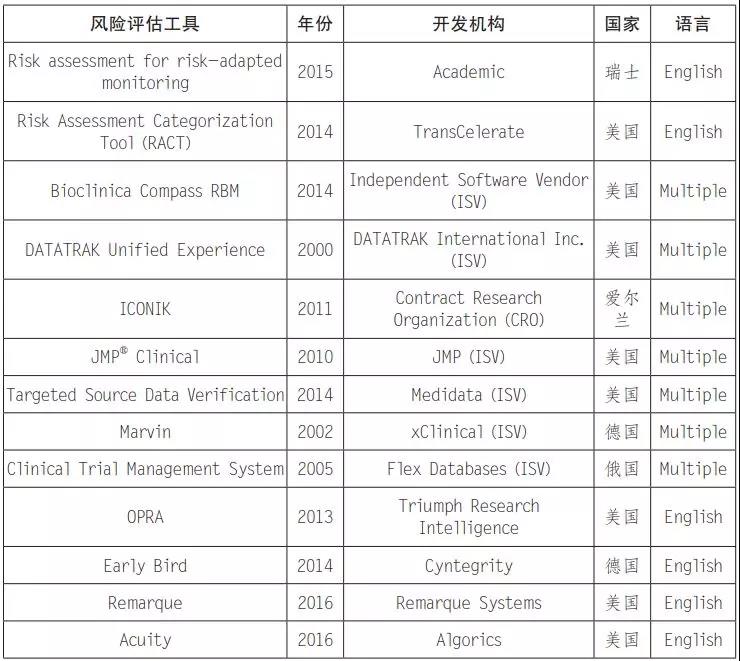
入组早期即在不同中心发生相似非预期不良事件,提示试验器械风险增加,需要申办者及时调整使用方法或终止试验。(见表 1)
表 1 适用于医疗器械临床试验的风险评估工具[5]
五、基于风险的医疗器械监管
自从 FDA 首次发布基于风险的临床试验监察指导原则距今已有 4 年时间,市场上已经出现多种临床试验专用风险分析软件(见表 1),证明此方法已经逐渐被行业和监管机构认可。基于风险的监察理念本身包含着大数据运用,结果导向以及个性管理等现代化方法,同样适用于监管机构临床试验核查、上市前审评审批以及上市后产品全生命周期持续监管。此文在此介绍临床试验监察方法的改革,亦是希望引发对于产品上市前审批方法的思考,强大的产业离不开有效的监管,有效的监管离不开监管科技的创新,在大数据爆发的时代,应尽快建立医疗器械产品标准数据库、质控数据库、常用材料可提取物数据库、危害风险评估预警、不良反应监测预警以及其他基于风险的医疗器械评价预警机制、分类风险评价机制,运用人工智能、数据与风险结合的方式建设基于多元数据库的监测 - 预警 - 评价 - 检查 - 控制为一体的监管平台,降低企业研发成本,加速我国医药行业的健康高质量发展。
|

